|
|

|
|
Molecular Mechanisms and Clinical Pathophysiology
of Maturity-Onset Diabetes of the Young
Stefan S. Fajans, M.D., Graeme I. Bell, Ph.D., and Kenneth
S. Polonsky, M.D.
|
|
Maturity-onset diabetes of the young (MODY) is a clinically heterogeneous
group of disorders characterized by nonketotic diabetes mellitus, an
autosomal dominant mode of inheritance, an onset usually before the
age of 25 years and frequently in childhood or adolescence, and a
primary defect in the function of the beta cells of the pancreas.
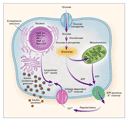
MODY can result from mutations in any one of at least six different
genes (Table 1).
One of these genes encodes the glycolytic enzyme glucokinase
(associated with MODY 2),3 and
the other five encode transcription factors: hepatocyte nuclear
factor (HNF) 4![]() (associated with MODY
1),4
HNF-1
(associated with MODY
1),4
HNF-1![]() (MODY 3),5
insulin promoter factor 1 (IPF-1 [MODY 4]),6
HNF-1
(MODY 3),5
insulin promoter factor 1 (IPF-1 [MODY 4]),6
HNF-1![]() (MODY 5),7 and
neurogenic differentiation factor 1 (NeuroD1), also known as
beta-cell E-box transactivator 2 (BETA2 [MODY 6]).8 All
these genes are expressed in beta cells, and mutation of any of them
leads to beta-cell dysfunction and diabetes mellitus (Figure 1). These
genes are also expressed in other tissues, and abnormalities in
liver and kidney function may also be evident in some forms of MODY.
Factors that affect insulin sensitivity, such as infection, puberty,
pregnancy, and (in rare cases) obesity, may trigger the onset of
diabetes and increase the severity of hyperglycemia in patients with
MODY, but otherwise, nongenetic factors have no important role in
the development of this disorder. Studies of MODY have led to a
better understanding of the genetic causes of beta-cell dysfunction.
(MODY 5),7 and
neurogenic differentiation factor 1 (NeuroD1), also known as
beta-cell E-box transactivator 2 (BETA2 [MODY 6]).8 All
these genes are expressed in beta cells, and mutation of any of them
leads to beta-cell dysfunction and diabetes mellitus (Figure 1). These
genes are also expressed in other tissues, and abnormalities in
liver and kidney function may also be evident in some forms of MODY.
Factors that affect insulin sensitivity, such as infection, puberty,
pregnancy, and (in rare cases) obesity, may trigger the onset of
diabetes and increase the severity of hyperglycemia in patients with
MODY, but otherwise, nongenetic factors have no important role in
the development of this disorder. Studies of MODY have led to a
better understanding of the genetic causes of beta-cell dysfunction.
|
|
Clinical Presentation
The most common clinical presentation of MODY is mild,
asymptomatic hyperglycemia in nonobese children, adolescents, and
young adults who have a prominent family history of diabetes, often
in successive generations (a pattern consistent with an autosomal
dominant mode of inheritance). Some patients have mild fasting
hyperglycemia for many years, whereas others have varying degrees of
glucose intolerance for several years before the onset of persistent
fasting hyperglycemia.9,10,11,12
Since mild hyperglycemia may not cause the classic symptoms of
diabetes, the diagnosis may not be made until adulthood. However,
according to prospective testing, it appears that in most patients
the onset is in childhood or adolescence. In some patients, there
may be rapid progression to overt asymptomatic or symptomatic
hyperglycemia, necessitating therapy with an oral hypoglycemic drug
or insulin.9,10,11,12,13
The presence of persistently normal plasma glucose concentrations in
persons with mutations in any of the known MODY-related genes is
unusual, and in the majority of them diabetes eventually develops
(with the exception of many patients with glucokinase mutations, as
discussed below). According to current estimates, MODY may account
for 1 to 5 percent of all cases of diabetes in the United States and
other industrialized countries.3,14
Several clinical characteristics distinguish patients with MODY
from those with type 2 diabetes, including a prominent family history
of diabetes in three or more generations, a young age at
presentation, and the absence of obesity (Table 2). In
recent years, type 2 diabetes has been recognized with increasing
frequency in adolescents who are obese, as are most older patients
with type 2 diabetes.
|
Function of the Gene
Products Implicated in MODY
Glucokinase
Glucokinase is expressed at its highest concentrations in
pancreatic beta cells and in the liver. It catalyzes the transfer of
phosphate from ATP to glucose, generating glucose-6-phosphate (Figure 1). This
reaction is the first, rate-limiting step in glucose metabolism.
Glucokinase functions as the glucose sensor in beta cells by
controlling the rate of entry of glucose into the glycolytic pathway
(glucose phosphorylation) and by controlling the rate of its
subsequent metabolism.16
The glucokinase that is expressed in the liver plays a key part in
the ability of that organ to store glucose as glycogen, particularly
in the postprandial state. Heterozygous mutations in the gene
encoding glucokinase lead to a partial deficiency of this enzyme and
are associated with MODY 2; homozygous mutations result in a
complete deficiency of this enzyme and lead to permanent neonatal
diabetes mellitus.17
HNF-1![]() , HNF-1
, HNF-1![]() ,
and HNF-4
,
and HNF-4![]()
The liver-enriched transcription factors HNF-1![]() , HNF-1
, HNF-1![]() , and HNF-4
, and HNF-4![]() were first discovered in studies designed to identify
the proteins responsible for the tissue-specific regulation of gene
expression in the liver.18
They are also found in other tissues and organs, including the
pancreatic islets, the kidneys, and genital tissues. HNF-1
were first discovered in studies designed to identify
the proteins responsible for the tissue-specific regulation of gene
expression in the liver.18
They are also found in other tissues and organs, including the
pancreatic islets, the kidneys, and genital tissues. HNF-1![]() and HNF-1
and HNF-1![]() are members of a family of transcription factors.18
HNF-4
are members of a family of transcription factors.18
HNF-4![]() is an orphan nuclear
receptor.19
HNF-1
is an orphan nuclear
receptor.19
HNF-1![]() , HNF-1
, HNF-1![]() , and HNF-4
, and HNF-4![]() constitute
part of a network of transcription factors that function together to
control gene expression during embryonic development and during adulthood
in tissues in which they are coexpressed. In pancreatic beta cells,
these transcription factors regulate the expression of the insulin
gene as well as the expression of genes encoding proteins involved
in glucose transport and metabolism20
and mitochondrial metabolism21 —
all of which are linked to insulin secretion. In the liver, these
proteins regulate lipoprotein biosynthesis.22
The expression of HNF-1
constitute
part of a network of transcription factors that function together to
control gene expression during embryonic development and during adulthood
in tissues in which they are coexpressed. In pancreatic beta cells,
these transcription factors regulate the expression of the insulin
gene as well as the expression of genes encoding proteins involved
in glucose transport and metabolism20
and mitochondrial metabolism21 —
all of which are linked to insulin secretion. In the liver, these
proteins regulate lipoprotein biosynthesis.22
The expression of HNF-1![]() is regulated
at least in part by HNF-4
is regulated
at least in part by HNF-4![]() .21
.21
IPF-1
IPF-1 is a transcription factor that was originally isolated as
a regulator of the transcription of the insulin and somatostatin genes.23
It also plays a central part in the development of the pancreas and
in the regulation of the expression of a variety of genes in the
pancreatic islets, including (in addition to the insulin gene) the
genes encoding glucokinase, islet amyloid polypeptide, and glucose
transporter 2.23
IPF-1 also appears to mediate glucose-induced stimulation of
insulin-gene transcription.24
NeuroD1 (BETA2)
The transcription factor NeuroD1, or BETA2, was isolated on the
basis of its ability to activate the transcription of the insulin
gene. It is required for the normal development of the pancreatic
islets.25
Clinical Features of Subtypes of MODY
MODY 2
Glucokinase-related MODY (MODY 2) is a common form of this
disorder, especially in children with mild hyperglycemia and in
women with gestational diabetes and a family history of diabetes.
It has been described in persons of all racial and ethnic groups.26
More than 130 MODY-associated mutations have been found in the glucokinase
gene. Heterozygous mutations in glucokinase are associated with a
mild form of nonprogressive hyperglycemia that is usually
asymptomatic at diagnosis and is treated with diet alone.27
The mild fasting hyperglycemia (blood glucose concentration, 110 to
145 mg per deciliter [6.1 to 8.0 mmol per liter]) and impaired
glucose tolerance in most affected carriers may be recognized by
biochemical testing at a young age, possibly as early as birth.3,28
About 50 percent of the women who are carriers may have gestational
diabetes.3,29
Less than 50 percent of the carriers have overt diabetes; of those
who do, many are obese or elderly. Two percent of carriers require insulin
therapy.30
Diabetes-associated complications are rare in this form of MODY.
The hyperglycemia in persons with glucokinase-related MODY appears
to result from a reduction in the sensitivity of beta cells to
glucose31
as well as a defect in postprandial glycogen synthesis in the liver.32
Heterozygous mutations in the gene encoding glucokinase are associated
with MODY and gestational diabetes. They are also associated with a
reduction in birth weight of 500 g or more, possibly because of
their effect on fetal insulin secretion.33
As noted above, homozygous mutations cause a complete deficiency of
glucokinase and are associated with permanent neonatal diabetes
mellitus, which is characterized by low birth weight and severe
diabetes, necessitating insulin treatment within the first few days
of life.17
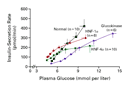
In patients with glucokinase-related MODY, the decrease in
glucokinase activity in the pancreatic beta cells leads to a
decrease in glucose phosphorylation in the beta cells, a reduction
in the sensitivity of beta cells to glucose, and a shift to the
right in the dose–response relation between the plasma glucose concentration
and insulin secretion (Figure 2 and Figure 3). These
effects occur across a broad range of glucose concentrations. There
is an increase in the threshold concentration of glucose necessary
to stimulate insulin secretion, from a normal basal concentration of
about 90 mg per deciliter (5.0 mmol per liter) to approximately 108
to 126 mg per deciliter (6.0 to 7.0 mmol per liter). As a result of
this upward shift in the plasma glucose threshold for stimulation of
insulin secretion, patients with glucokinase mutations have mildly
increased basal and postprandial plasma glucose concentrations (Figure 4).31
Surprisingly, in view of the key part played by glucokinase in the
sensitivity of beta cells to glucose and in insulin secretion, the
hyperglycemia in these patients is usually mild and does not
increase substantially over the course of many years.3,27
It appears that in patients with glucokinase mutations, physiologic
adaptation within the pancreatic beta cells limits the severity of
hyperglycemia.
|

|
|
This hypothesis was explored in a study of subjects with different types
of glucokinase mutations and normal subjects.35
In two subjects who had glucokinase mutations associated with only
a small decrease in enzymatic activity, the decrease in insulin secretion
was directly proportional to the decrease in the glucokinase-mediated flux
of glucose. However, in four subjects with glucokinase mutations
that resulted in severe decreases in enzymatic activity, the amount
of insulin secreted was less than that in normal subjects, but the
difference was smaller than predicted, suggesting the presence of
compensatory mechanisms within the pancreatic beta cells that
resulted in a relative increase in the insulin-secretion response.
The nature of this compensatory or adaptive mechanism has been
examined in mice with glucokinase-related MODY (mice with only one
glucokinase-gene allele).36
Islets from these mice and from control mice were incubated at
various glucose concentrations, and the findings suggested that mild
hyperglycemia leads to increased expression of the single wild-type
glucokinase-gene allele, thus limiting the severity of the defect in
glucose-stimulated insulin secretion. Since the hepatic glucokinase
promoter is regulated predominantly by insulin and not by glucose,
the compensatory mechanism is probably operative only in beta cells.
MODY 1 and MODY 3
Not unexpectedly, the pathophysiologic mechanisms of MODY due
to mutations in the HNF-4![]() gene (MODY 1) and MODY
due to mutations in the HNF-1
gene (MODY 1) and MODY
due to mutations in the HNF-1![]() gene
(MODY 3) are very similar, since HNF-4
gene
(MODY 3) are very similar, since HNF-4![]() regulates the expression of HNF-1
regulates the expression of HNF-1![]() . Like
persons with glucokinase mutations, those with mutations in the
HNF-4
. Like
persons with glucokinase mutations, those with mutations in the
HNF-4![]() gene or the HNF-1
gene or the HNF-1![]() gene may present
with a mild form of diabetes. Despite similarly mild elevations in
fasting plasma glucose concentrations, however, they have
significantly higher plasma glucose concentrations two hours after
glucose administration than do persons with glucokinase mutations.27
The hyperglycemia in patients with HNF-1
gene may present
with a mild form of diabetes. Despite similarly mild elevations in
fasting plasma glucose concentrations, however, they have
significantly higher plasma glucose concentrations two hours after
glucose administration than do persons with glucokinase mutations.27
The hyperglycemia in patients with HNF-1![]() –related or HNF-4
–related or HNF-4![]() –related MODY tends to
increase over time, resulting in the need for treatment with oral
hypoglycemic drugs or insulin in a substantial proportion of these
patients (30 to 40 percent require insulin).9,10,11,12,13
These forms of MODY are associated with a progressive decrease in
insulin secretion. For example, prospective studies in a large
family with HNF-4
–related MODY tends to
increase over time, resulting in the need for treatment with oral
hypoglycemic drugs or insulin in a substantial proportion of these
patients (30 to 40 percent require insulin).9,10,11,12,13
These forms of MODY are associated with a progressive decrease in
insulin secretion. For example, prospective studies in a large
family with HNF-4![]() –related MODY revealed
that glucose-induced insulin secretion decreased at a rate of 1 to 4
percent per year.12
This progression suggests that the beta cells are not able to
compensate for the deficiency of HNF-4
–related MODY revealed
that glucose-induced insulin secretion decreased at a rate of 1 to 4
percent per year.12
This progression suggests that the beta cells are not able to
compensate for the deficiency of HNF-4![]() .
.
In most populations, mutations in the HNF-1![]() gene (resulting in MODY 3) are the most common cause
of MODY. To date, more than 120 mutations in this gene have been
identified in persons of all racial and ethnic backgrounds — for
example, European, Chinese, Japanese, African, and American Indian37 (and
unpublished data). Mutations in the HNF-1
gene (resulting in MODY 3) are the most common cause
of MODY. To date, more than 120 mutations in this gene have been
identified in persons of all racial and ethnic backgrounds — for
example, European, Chinese, Japanese, African, and American Indian37 (and
unpublished data). Mutations in the HNF-1![]() gene appear to be the most common cause of MODY among
adults seen in diabetes clinics. In contrast, mutations in the HNF-4
gene appear to be the most common cause of MODY among
adults seen in diabetes clinics. In contrast, mutations in the HNF-4![]() gene (resulting in MODY
1) are relatively uncommon; to date, only 13 families worldwide have
been identified as having this form of MODY.
gene (resulting in MODY
1) are relatively uncommon; to date, only 13 families worldwide have
been identified as having this form of MODY.
Patients with HNF-1![]() –related or HNF-4
–related or HNF-4![]() –related MODY may
have the full spectrum of complications of diabetes. Microvascular complications,
particularly those involving the retinas and kidneys, are as common
in these patients as in patients with type 1 or type 2 diabetes
(matched according to the duration of diabetes and the degree of
glycemic control) and are thus probably determined by the degree of
glycemic control (Table
1). 10,30,38,39
–related MODY may
have the full spectrum of complications of diabetes. Microvascular complications,
particularly those involving the retinas and kidneys, are as common
in these patients as in patients with type 1 or type 2 diabetes
(matched according to the duration of diabetes and the degree of
glycemic control) and are thus probably determined by the degree of
glycemic control (Table
1). 10,30,38,39
Studies performed in prediabetic carriers of HNF-4![]() and HNF-1
and HNF-1![]() mutations
showed that these two groups had similar defects in the pattern of
glucose-induced insulin secretion, without impairment of insulin
sensitivity. Thus, impaired beta-cell function, rather than a defect
in insulin activity, appears to be the primary cause of diabetes in
persons with these two forms of MODY.40,41,42,43
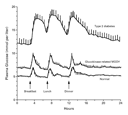
After an overnight fast, insulin secretion is normal. However, as
plasma glucose concentrations increase to approximately 125 to 145
mg per deciliter (7.0 to 8.0 mmol per liter), insulin secretion does
not continue to increase, as it would in nondiabetic persons, but
rather begins to level off (Figure 3).41,42
It is possible to distinguish persons with an HNF-1
mutations
showed that these two groups had similar defects in the pattern of
glucose-induced insulin secretion, without impairment of insulin
sensitivity. Thus, impaired beta-cell function, rather than a defect
in insulin activity, appears to be the primary cause of diabetes in
persons with these two forms of MODY.40,41,42,43
After an overnight fast, insulin secretion is normal. However, as
plasma glucose concentrations increase to approximately 125 to 145
mg per deciliter (7.0 to 8.0 mmol per liter), insulin secretion does
not continue to increase, as it would in nondiabetic persons, but
rather begins to level off (Figure 3).41,42
It is possible to distinguish persons with an HNF-1![]() mutation from those with an HNF-4
mutation from those with an HNF-4![]() mutation according to the ability of glucose to prime
the insulin-secretion response to a subsequent glucose stimulus. In
the prediabetic state, the normal priming effect of mild
hyperglycemia on insulin secretion is retained in persons with HNF-1
mutation according to the ability of glucose to prime
the insulin-secretion response to a subsequent glucose stimulus. In
the prediabetic state, the normal priming effect of mild
hyperglycemia on insulin secretion is retained in persons with HNF-1![]() mutations (as it is in
those with glucokinase mutations or no mutations), but it is lost in
those with HNF-4
mutations (as it is in
those with glucokinase mutations or no mutations), but it is lost in
those with HNF-4![]() mutations.
mutations.
Both prediabetic and diabetic persons with mutations in the HNF-4![]() gene secrete decreased
amounts of insulin in response to glucose and in response to
arginine and also have an impairment of glucagon secretion in
response to arginine.44
Impairment of glucagon secretion in response to arginine has also
been reported in a patient with diabetes due to a mutation in the
HNF-1
gene secrete decreased
amounts of insulin in response to glucose and in response to
arginine and also have an impairment of glucagon secretion in
response to arginine.44
Impairment of glucagon secretion in response to arginine has also
been reported in a patient with diabetes due to a mutation in the
HNF-1![]() gene (MODY 3).45
Furthermore, a defect in the hypoglycemia-induced secretion of
pancreatic polypeptide has been found in prediabetic and diabetic
persons who have mutations in the gene for HNF-4
gene (MODY 3).45
Furthermore, a defect in the hypoglycemia-induced secretion of
pancreatic polypeptide has been found in prediabetic and diabetic
persons who have mutations in the gene for HNF-4![]() .46
These findings suggest that a deficiency of HNF-4
.46
These findings suggest that a deficiency of HNF-4![]() activity resulting from mutations in this gene may
affect the function of the beta, alpha, and pancreatic polypeptide
cells of the pancreatic islets.
activity resulting from mutations in this gene may
affect the function of the beta, alpha, and pancreatic polypeptide
cells of the pancreatic islets.
To examine the molecular basis of the defect in insulin secretion
resulting from decreased activity of HNF-1![]() , we
have studied mice that lack this gene. These animals have marked
hyperglycemia and defects in glucose-induced insulin secretion as a
result of defective beta-cell glycolytic signaling.47,48
The consequences of HNF-4
, we
have studied mice that lack this gene. These animals have marked
hyperglycemia and defects in glucose-induced insulin secretion as a
result of defective beta-cell glycolytic signaling.47,48
The consequences of HNF-4![]() deficiency have also been studied in embryonic stem cells,20
and the results show that HNF-4
deficiency have also been studied in embryonic stem cells,20
and the results show that HNF-4![]() regulates the
expression of proteins involved in glucose transport and glycolysis,
which are required for a normal, glucose-dependent insulin secretory
response. In the INS-1 cell line, defective HNF-4
regulates the
expression of proteins involved in glucose transport and glycolysis,
which are required for a normal, glucose-dependent insulin secretory
response. In the INS-1 cell line, defective HNF-4![]() –dependent insulin secretion is linked to impaired
mitochondrial metabolism.21
–dependent insulin secretion is linked to impaired
mitochondrial metabolism.21
In addition to their effects on beta-cell function, deficiencies
of HNF-1![]() and HNF-4
and HNF-4![]() affect kidney and liver function, respectively. Patients
with HNF-1
affect kidney and liver function, respectively. Patients
with HNF-1![]() mutations have
decreased renal reabsorption of glucose (i.e., a low renal threshold
for glucose) and glycosuria.49,50
A deficiency of HNF-4
mutations have
decreased renal reabsorption of glucose (i.e., a low renal threshold
for glucose) and glycosuria.49,50
A deficiency of HNF-4![]() affects triglyceride
and apolipoprotein biosynthesis and is associated with a 50 percent
reduction in serum triglyceride concentrations and a 25 percent
reduction in serum concentrations of apolipoproteins AII and CIII
and Lp(a) lipoprotein.51,52
affects triglyceride
and apolipoprotein biosynthesis and is associated with a 50 percent
reduction in serum triglyceride concentrations and a 25 percent
reduction in serum concentrations of apolipoproteins AII and CIII
and Lp(a) lipoprotein.51,52
MODY 4
Mutations in the gene that encodes IPF-1 are a rare cause of MODY;
in fact, the current understanding of this form of MODY (MODY 4) is
based on studies of a single family. The proband was an infant with
permanent neonatal diabetes and pancreatic exocrine insufficiency
resulting from congenital agenesis of the pancreas.53
Molecular genetic studies revealed that this infant was homozygous
for a frame-shift mutation in the gene for IPF-1 and that both
parents were heterozygous for this mutation.6
Studies of the extended family revealed a high prevalence of a
mild form of diabetes that had an autosomal dominant pattern of
inheritance and was associated with the heterozygous mutation in
IPF-1. The expression of diabetes in this pedigree may occur at
later ages than in families with other types of MODY. Six diabetic
family members who were heterozygous for the mutation (and whose
mean fasting plasma glucose concentration was 169 mg per deciliter
[9.4 mmol per liter]) had severe impairment of insulin secretion
during a hyperglycemic clamp study; no such impairment was seen in
five family members who did not have the mutation.54
MODY 5
Mutations in the gene encoding HNF-1![]() are the cause of an uncommon but distinct form of MODY
(MODY 5) that is characterized by both diabetes mellitus and renal
cysts (hypoplastic glomerulocystic kidney disease).7,55,56,57,58
In addition, two of four female carriers in one family had internal
genital abnormalities (vaginal aplasia and a rudimentary uterus),56
and in another family a female carrier had a bicornuate uterus.57
Thus, heterozygous mutations in the gene for HNF-1
are the cause of an uncommon but distinct form of MODY
(MODY 5) that is characterized by both diabetes mellitus and renal
cysts (hypoplastic glomerulocystic kidney disease).7,55,56,57,58
In addition, two of four female carriers in one family had internal
genital abnormalities (vaginal aplasia and a rudimentary uterus),56
and in another family a female carrier had a bicornuate uterus.57
Thus, heterozygous mutations in the gene for HNF-1![]() may be associated with a spectrum of clinical
features, which may be determined by the nature of the specific
mutation and its effect on HNF-1
may be associated with a spectrum of clinical
features, which may be determined by the nature of the specific
mutation and its effect on HNF-1![]() function.
function.
MODY Associated with Mutations in Other
Genes
The identification of mutations in the genes encoding glucokinase
and transcription factors HNF-4![]() ,
HNF-1
,
HNF-1![]() , HNF-1
, HNF-1![]() , and IPF-1 suggests that MODY is a disorder involving
abnormal gene expression, abnormal glucose metabolism, or both in
the beta cells. This possibility has led investigators to screen for
mutations in other genes in these pathways, especially those
encoding transcription factors expressed in beta cells, in families
with MODY or autosomal dominant forms of type 2 diabetes.
, and IPF-1 suggests that MODY is a disorder involving
abnormal gene expression, abnormal glucose metabolism, or both in
the beta cells. This possibility has led investigators to screen for
mutations in other genes in these pathways, especially those
encoding transcription factors expressed in beta cells, in families
with MODY or autosomal dominant forms of type 2 diabetes.
Mutations in the gene encoding transcription factor NeuroD1 (BETA2)
were found in two families with autosomal dominant type 2 diabetes.8 One
of these families met the criteria for MODY, including (in addition
to an autosomal dominant pattern of inheritance) an onset of
diabetes before 25 years of age in three carriers and a requirement
for insulin treatment — a finding consistent with the presence of
beta-cell dysfunction — in five carriers. Thus, mutations in NeuroD1
may be the cause of another subtype of MODY, designated MODY 6.
A nonsense mutation was found in the gene encoding beta-cell transcription
factor Islet-1 in a Japanese family.59
This mutation led to decreased activity of this transcription factor
and thus may have been pathogenic. However, additional genetic and
clinical studies are required to determine whether mutations in
Islet-1 are the cause of another subtype of MODY.
Families whose members have a clinical history compatible with
a diagnosis of MODY and who do not have mutations in any of the
six known MODY-related genes account for an estimated 15 to 20
percent of European persons who have clinical MODY and as many as 80
percent of Japanese persons with clinical MODY.37
We believe that, in time, additional MODY-related genes will be
identified and that they will explain the molecular basis of
diabetes in these patients.
Mutations in MODY-Related Genes in Type 2
Diabetes
It does not appear that mutations in known MODY-related genes
contribute to the development of hyperglycemia in the majority of
patients with type 2 diabetes, although mutations have been found in
a few patients.60,61,62,63
One group in which a mutation in a MODY-associated gene, the gene
encoding HNF-1![]() , may be a key
genetic factor linked to type 2 diabetes is the Canadian Oji-Cree
Indians, who live in northwestern Ontario and Manitoba.64
Thus, sequence variation in MODY-related genes may contribute to
the polygenic background and to the development of type 2 diabetes (Table 2). The
absence of an association with type 2 diabetes in one population
should not deter investigators from studying these genes in other
populations.
, may be a key
genetic factor linked to type 2 diabetes is the Canadian Oji-Cree
Indians, who live in northwestern Ontario and Manitoba.64
Thus, sequence variation in MODY-related genes may contribute to
the polygenic background and to the development of type 2 diabetes (Table 2). The
absence of an association with type 2 diabetes in one population
should not deter investigators from studying these genes in other
populations.
Despite the lack of evidence that mutations in MODY-related genes
are responsible for the development of type 2 diabetes in the
majority of patients with this disorder, the mechanisms by which these
mutations lead to hyperglycemia may be relevant to the
pathophysiology of the beta-cell defects in type 2 diabetes. Defects
in the metabolism of glucose in pancreatic beta cells may be the
mechanism by which insulin secretion is impaired in type 2 diabetes.
Thus, lessons learned from studies of MODY may improve our
understanding of the insulin insufficiency associated with type 2
diabetes.
Genetic Screening for MODY
With the identification of genes responsible for MODY, it is possible
to identify members of pedigrees who have inherited the specific
mutation affecting their family, even before carbohydrate intolerance
develops. Indeed, in the past few years, some parents have requested
that their children undergo genetic screening for the mutation in their
family.65
If a child does not carry the mutation, further clinical testing is
unnecessary. If a child does carry the mutation, periodic testing
for slight abnormalities of carbohydrate metabolism is recommended.
These principles can be applied to any family with MODY for which
the mutation is known.
Genetic screening for and identification of a specific
MODY-related mutation in children may have important prognostic and
therapeutic implications. If carbohydrate intolerance or diabetes is
due to a mutation in the gene encoding glucokinase, limited therapy
and follow-up are adequate, because of the benign and nonprogressive
course of diabetes in such cases. On the other hand, persons who
are genetically susceptible to diabetes due to mutations in the
genes for HNF-1![]() and HNF-4
and HNF-4![]() should be monitored frequently so that appropriate
therapy can be instituted early in the course of their
hyperglycemia, because of the risk of progression to severe
hyperglycemia and insulin-requiring diabetes. Early treatment to
achieve normoglycemia should prevent vascular and neuropathic complications.
Thus, MODY is one type of diabetes that warrants genetic counseling,
because of the known mode of inheritance and high penetrance.
should be monitored frequently so that appropriate
therapy can be instituted early in the course of their
hyperglycemia, because of the risk of progression to severe
hyperglycemia and insulin-requiring diabetes. Early treatment to
achieve normoglycemia should prevent vascular and neuropathic complications.
Thus, MODY is one type of diabetes that warrants genetic counseling,
because of the known mode of inheritance and high penetrance.
Genetic diagnosis may also be advised for patients who have been
classified as having type 1 diabetes and who have a strong family
history of diabetes. An appreciable fraction of these patients have
been found to carry the HNF-1![]() mutation.66,67,68
The diagnosis of HNF-1
mutation.66,67,68
The diagnosis of HNF-1![]() –related MODY rather
than type 1 diabetes has implications for prognosis in these
patients. Unfortunately, these approaches are currently applicable
only in a research setting; commercial tests for the identification
of MODY-related genes are not yet available.
–related MODY rather
than type 1 diabetes has implications for prognosis in these
patients. Unfortunately, these approaches are currently applicable
only in a research setting; commercial tests for the identification
of MODY-related genes are not yet available.
Conclusions
A better understanding of the causes and pathophysiology of MODY
is emerging from genetic, molecular biologic, and physiological studies
of this disorder. We believe this knowledge will lead to new
therapeutic approaches and agents that will prevent, correct, or at
least delay the decline in pancreatic beta-cell function that
characterizes not only MODY but also type 2 diabetes.
Supported by grants from the National Institutes of Health
(DK-20572 and DK-20595, to the Diabetes Research and Training
Centers at the University of Michigan and the University of Chicago,
respectively; RR-00042 and RR-00055, to the General Clinical Research
Centers at the University of Michigan and the University of Chicago,
respectively; DK-31842; and DK-44840), by a gift from the
Blum–Kovler Foundation, and by funds from the Howard Hughes Medical
Institute.
We are indebted to Dr. Franz Matschinsky for valuable discussions
about glucokinase mutations.
Source Information
From the Department of Internal Medicine, Division of
Endocrinology and Metabolism, University of Michigan Health System, Ann Arbor
(S.S.F.); the Howard Hughes Medical Institute and Departments of Biochemistry
and Molecular Biology, Medicine, and Human Genetics, University of Chicago,
Chicago (G.I.B.); and the Departments of Medicine and Cell Biology and
Physiology, Washington University School of Medicine, St. Louis (K.S.P.).
Address reprint requests to Dr. Fajans at 3920 Taubman Ctr., Box
0354, University Hospitals, Ann Arbor, MI 48109-0354, or at [log in to unmask].
References
- Søvik
O, Njølstad P, Følling I, Sagen J, Cockburn BN, Bell GI. Hyperexcitability
to sulphonylurea in MODY3. Diabetologia 1998;41:607-608.
[Medline]
- Pearson
ER, Liddell WG, Shepherd M, Corrall RJ, Hattersley AT. Sensitivity to sulphonylureas
in patients with hepatocyte nuclear factor-1
 gene mutations: evidence for pharmacogenetics in diabetes.
Diabet Med 2000;17:543-545.
gene mutations: evidence for pharmacogenetics in diabetes.
Diabet Med 2000;17:543-545.[Medline]
- Froguel
P, Zouali H, Vionnet N, et al. Familial hyperglycemia due to mutations in
glucokinase: definition of a subtype of diabetes mellitus. N Engl J Med
1993;328:697-702.
[Abstract/Full Text]
- Yagamata
K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor-4 gene in
maturity-onset diabetes of the young (MODY1). Nature 1996;384:458-460.
[Medline]
- Yamagata
K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1 gene in
maturity-onset diabetes of the young (MODY3). Nature 1996;384:455-458.
[Medline]
- Stoffers
DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus
(MODY4) linked to IPF1. Nat Genet 1997;17:138-139.
[Medline]
- Horikawa
Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1
 gene (TCF2)
associated with MODY. Nat Genet 1997;17:384-385.
gene (TCF2)
associated with MODY. Nat Genet 1997;17:384-385.[Medline]
- Malecki
MT, Jhala US, Antonellis A, et al. Mutations in NEUROD1 are associated
with the development of type 2 diabetes mellitus. Nat Genet
1999;23:323-328.
[Medline]
- Fajans
SS. MODY: a model for understanding the pathogeneses and natural history
of type II diabetes. Horm Metab Res 1987;19:591-599.
[Medline]
- Fajans
SS. Scope and heterogeneous nature of MODY. Diabetes Care 1990;13:49-64.
[Erratum, Diabetes Care 1990;13:910.]
[Medline]
- Fajans
SS, Bell GI, Bowden DW, Halter JB, Polonsky KS. Maturity-onset diabetes of
the young. Life Sci 1994;55:413-422.
[Medline]
- Fajans
SS, Brown MB. Administration of sulfonylureas can increase glucose-induced
insulin secretion for decades in patients with maturity-onset diabetes of
the young. Diabetes Care 1993;16:1254-1261.
[Medline]
- Frayling
TM, Bulman MP, Ellard S, et al. Mutations in the hepatocyte nuclear
factor-1 gene are a common
cause of maturity-onset diabetes of the young in the U.K. Diabetes
1997;46:720-725.
[Medline]
- Ledermann
HM. Maturity-onset diabetes of the young (MODY) at least ten times more
common in Europe than previously assumed? Diabetologia 1995;38:1482-1482.
- Fajans
SS, Bell GI, Bowden DW. MODY: a model for the study of the molecular
genetics of NIDDM. J Lab Clin Med 1992;119:206-210.
[Medline]
- Matschinsky
FM, Glaser B, Magnuson MA. Pancreatic -cell
glucokinase: closing the gap between theoretical concepts and experimental
realities. Diabetes 1998;47:307-315.
[Medline]
- Njølstad
PR, Søvik O, Cuesta-Muñoz A, et al. Neonatal diabetes mellitus due to
complete glucokinase deficiency. N Engl J Med 2001;344:1588-1592.
[Full Text]
- Cereghini
S. Liver-enriched transcription factors and hepatocyte differentiation.
FASEB J 1996;10:267-282.
[Abstract]
- Duncan
SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a
transcription factor network required for differentiation and metabolism.
Science 1998;281:692-695.
[Abstract/Full Text]
- Stoffel
M, Duncan SA. The maturity-onset diabetes of the young (MODY1)
transcription factor HNF4 regulates
expression of genes required for glucose transport and metabolism. Proc
Natl Acad Sci U S A 1997;94:13209-13214.
[Abstract/Full Text]
- Wang H,
Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear
factor 4 regulates the
expression of pancreatic -cell genes
implicated in glucose metabolism and nutrient-induced insulin secretion. J
Biol Chem 2000;275:35953-35959.
[Abstract/Full Text]
- Ladias
JA, Hadzopoulou-Cladaras M, Kardassis D, et al. Transcriptional regulation
of human apolipoprotein genes ApoB, ApoCIII, and ApoAII by members of the
steroid hormone receptor superfamily HNF-4, ARP-1, EAR-2, and EAR-3. J
Biol Chem 1992;267:15849-15860.
[Abstract]
- St-Onge
L, Wehr R, Gruss P. Pancreas development and diabetes. Curr Opin Genet Dev
1999;9:295-300.
[Medline]
- Marshak
S, Totary H, Cerasi E, Melloul D. Purification of the -cell
glucose-sensitive factor that transactivates the insulin gene
differentially in normal and transformed islet cells. Proc Natl Acad Sci U
S A 1996;93:15057-15062.
[Abstract/Full Text]
- Naya FJ,
Huang HP, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and
abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice.
Genes Dev 1997;11:2323-2334.
[Abstract/Full Text]
- Bell GI,
Cuesta-Munoz A, Matschinsky FM. Glucokinase. In: Creighton T, ed.
Encyclopedia of molecular medicine. New York: John Wiley (in press).
- Pearson
ER, Velho G, Clark P, et al. -Cell
genes and diabetes: quantitative and qualitative differences in the
pathophysiology of hepatic nuclear factor-1
and glucokinase mutations. Diabetes 2001;50:S101-S107.
[Medline]
- Prisco
F, Iafusco D, Franzese A, Sulli N, Barbetti F. MODY2 presenting as
neonatal hyperglycaemia: a need to reshape the definition of
"neonatal diabetes"? Diabetologia 2000;43:1331-1332.
[Medline]
- Ellard
S, Beards F, Allen LI, et al. A high prevalence of glucokinase mutations
in gestational diabetic subjects selected by clinical criteria.
Diabetologia 2000;43:250-253.
[Medline]
- Velho G,
Vaxillaire M, Boccio V, Charpentier G, Froguel P. Diabetes complications
in NIDDM kindreds linked to the MODY3 locus on chromosome 12q. Diabetes
Care 1996;19:915-919.
[Medline]
- Byrne
MM, Sturis J, Clément K, et al. Insulin secretory abnormalities in
subjects with hyperglycemia due to glucokinase mutations. J Clin Invest
1994;93:1120-1130.
[Medline]
- Velho G,
Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in
glucokinase-deficient (MODY-2) subjects. J Clin Invest 1996;98:1755-1761.
[Abstract/Full Text]
- Hattersley
AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S. Mutations in
the glucokinase gene of the fetus result in reduced birth weight. Nat
Genet 1998;19:268-270.
[Medline]
- Polonsky
KS. The -cell in diabetes:
from molecular genetics to clinical research. Diabetes 1995;44:705-717.
[Medline]
- Sturis
J, Kurland IJ, Byrne MM, et al. Compensation in pancreatic beta-cell
function in subjects with glucokinase mutations. Diabetes 1994;43:718-723.
[Medline]
- Sreenan
SK, Cockburn BN, Baldwin AC, et al. Adaptation to hyperglycemia enhances
insulin secretion in glucokinase mutant mice. Diabetes 1998;47:1881-1888.
[Medline]
- Fajans
SS, Bell GI. Maturity-onset diabetes of the young: a model for genetic
studies of diabetes mellitus: In: LeRoith D, Taylor SI, Olefsky JM, eds.
Diabetes mellitus: a fundamental and clinical text. 2nd ed. Philadelphia:
Lippincott Williams & Wilkins, 2000:691-705.
- Isomaa
B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients
with MODY3 diabetes. Diabetologia 1998;41:467-473.
[Medline]
- Iwasaki
N, Ogata M, Tomonaga O, et al. Liver and kidney function in Japanese
patients with maturity-onset diabetes of the young. Diabetes Care
1998;21:2144-2148.
[Medline]
- Herman
WH, Fajans SS, Ortiz FJ, et al. Abnormal insulin secretion, not insulin
resistance, is the genetic or primary defect of MODY in the RW pedigree.
Diabetes 1994;43:40-46. [Erratum, Diabetes 1994;43:1171.]
[Medline]
- Byrne
MM, Sturis J, Fajans SS, et al. Altered insulin secretory responses to
glucose in subjects with a mutation in the MODY1 gene on chromosome 20.
Diabetes 1995;44:699-704.
[Medline]
- Byrne
MM, Sturis J, Menzel S, et al. Altered insulin secretory responses to
glucose in diabetic and nondiabetic subjects with mutations in the
diabetes susceptibility gene MODY3 on chromosome 12. Diabetes
1996;45:1503-1510.
[Medline]
- Lehto M,
Tuomi T, Mahtani MM, et al. Characterization of the MODY3 phenotype:
early-onset diabetes caused by an insulin secretion defect. J Clin Invest
1997;99:582-591.
[Abstract/Full Text]
- Herman
WH, Fajans SS, Smith MJ, Polonsky KS, Bell GI, Halter JB. Diminished
insulin and glucagon secretory responses to arginine in nondiabetic subjects
with a mutation in the hepatocyte nuclear factor-4/MODY1 gene. Diabetes 1997;46:1749-1754.
[Medline]
- Yoshiuchi
I, Yamagata K, Yang Q, et al. Three new mutations in the hepatocyte
nuclear factor-1 gene in Japanese
subjects with diabetes mellitus: clinical features and functional
characterization. Diabetologia 1999;42:621-626.
[Medline]
- Ilag LL,
Tabaei BP, Herman WH, et al. Reduced pancreatic polypeptide response to
hypoglycemia and amylin response to arginine in subjects with a mutation
in the HNF-4/MODY1 gene.
Diabetes 2000;49:961-968.
[Medline]
- Pontoglio
M, Sreenan S, Roe M, et al. Defective insulin secretion in hepatocyte
nuclear factor 1-deficient mice. J
Clin Invest 1998;101:2215-2222.
[Abstract/Full Text]
- Dukes
ID, Sreenan S, Roe MW, et al. Defective pancreatic -cell glycolytic signaling in hepatocyte nuclear factor-1-deficient mice. J
Biol Chem 1998;273:24457-24464.
[Abstract/Full Text]
- Menzel
R, Kaisaki PJ, Rjasanowski I, Heinke P, Kerner W, Menzel S. A low renal
threshold for glucose in diabetic patients with a mutation in the
hepatocyte nuclear factor-1 (HNF-1) gene. Diabet Med
1998;15:816-820.
[Medline]
- Pontoglio
M, Prié D, Cheret C, et al. HNF1
controls renal glucose reabsorption in mouse and man. EMBO Rep
2000;4:359-365.
- Shih DQ,
Dansky HM, Fleisher M, Assmann G, Fajans SS, Stoffel M. Genotype/phenotype
relationships in HNF-4/MODY1:
haploinsufficiency is associated with reduced apolipoprotein (AII),
apolipoprotein (CIII), lipoprotein(a), and triglyceride levels. Diabetes
2000;49:832-837.
[Medline]
- Lehto M,
Bitzen PO, Isomaa B, et al. Mutation in the HNF-4 gene affects insulin secretion and triglyceride metabolism.
Diabetes 1999;48:423-425.
[Medline]
- Wright
NM, Metzger DL, Borowitz SM, Clarke WL. Permanent neonatal diabetes
mellitus and pancreatic exocrine insufficiency resulting from congenital
pancreatic agenesis. Am J Dis Child 1993;147:607-609.
- Clocquet
AR, Egan JM, Stoffers DA, et al. Impaired insulin secretion and increased
insulin sensitivity in familial maturity-onset diabetes of the young 4
(insulin promoter factor 1 gene). Diabetes 2000;49:1856-1864.
[Medline]
- Nishigori
H, Yamada S, Kohama T, et al. Frameshift mutation, A263fsinsGG, in the
hepatocyte nuclear factor-1
gene associated with diabetes and renal dysfunction. Diabetes
1998;47:1354-1355.
[Medline]
- Lindner
TH, Njølstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome
of diabetes mellitus, renal dysfunction and genital malformation
associated with a partial deletion of the pseudo-POU domain of hepatocyte
nuclear factor-1. Hum Mol Genet
1999;8:2001-2008.
[Abstract/Full Text]
- Iwasaki
N, Okabe I, Momoi MY, Ohashi H, Ogata M, Iwamoto Y. Splice site mutation
in the hepatocyte nuclear factor-1
gene, IVS2nt+1G>A, associated with maturity-onset diabetes of the
young, renal dysplasia and bicornuate uterus. Diabetologia 2001;44:387-388.
[Medline]
- Bingham
C, Bulman MP, Ellard S, et al. Mutations in the hepatocyte nuclear
factor-1 gene are associated
with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet
2001;68:219-224.
[Medline]
- Shimomura
H, Sanke T, Hanabusa T, Tsunoda K, Furata H, Nanjo K. Nonsense mutation of
islet-1 gene (Q310X) found in a type 2 diabetic patient with a strong
family history. Diabetes 2000;49:1597-1600.
[Medline]
- Hani EH,
Stoffers DA, Chèvre JC, et al. Defective mutations in the insulin promoter
factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin
Invest 1999;104:R41-R48.
[Medline]
- Macfarlane
WM, Frayling TM, Ellard S, et al. Missense mutations in the insulin
promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest
1999;104:R33-R39.
[Medline]
- Iwasaki
N, Oda N, Ogata M, et al. Mutations in the hepatocyte nuclear factor-1/MODY3 gene in
Japanese subjects with early- and late-onset NIDDM. Diabetes 1997;46:1504-1508.
[Medline]
- Hani EH,
Suaud L, Boutin P, et al. A missense mutation in hepatocyte nuclear
factor-4, resulting in a
reduced transactivation activity, in human late-onset
non-insulin-dependent diabetes mellitus. J Clin Invest 1998;101:521-526.
[Abstract/Full Text]
- Hegele
RA, Cao H, Harris SB, Hanley AJ, Zinman B. The hepatic nuclear factor-1 G319S variant is
associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin
Endocrinol Metab 1999;84:1077-1082.
[Abstract/Full Text]
- Fajans
SS, Bell GI, Herman WH, et al. Natural history, genetics and pathogenesis
of HNF-4/MODY1: a 40-year
prospective study of the RW pedigree. In: Matschinsky FM, Magnuson MA,
eds. Vol. 15 of Frontiers in diabetes: molecular pathogenesis of MODYs.
Basel, Switzerland: Karger, 2000:1-15.
- Yamada
S, Nishigori H, Onda H, et al. Identification of mutations in the
hepatocyte nuclear factor (HNF)-1
gene in Japanese subjects with IDDM. Diabetes 1997;46:1643-1647.
[Medline]
- Møller
AM, Dalgaard LT, Pociot F, Nerup J, Hansen T, Pedersen O. Mutations in the
hepatocyte nuclear factor-1 gene in Caucasian
families originally classified as having type I diabetes. Diabetologia
1998;41:1528-1531.
[Medline]
- Hathout
EH, Cockburn BN, Mace JW, Sharkey J, Chen-Daniel J, Bell GI. A case of
hepatocyte nuclear factor-1 diabetes/MODY3
masquerading as type 1 diabetes in a Mexican-American adolescent and
responsive to a low dose of sulfonylurea. Diabetes Care 1999;22:867-868.
[Medline]
Edward E.
Rylander, M.D.
Diplomat American
Board of Family Practice.
Diplomat American
Board of Palliative Medicine.