|
|

|
|
Intravenous Zoledronic Acid in Postmenopausal
Women with Low Bone Mineral Density
Ian R. Reid, M.D., Jacques P. Brown, M.D., Peter Burckhardt,
M.D., Zebulun Horowitz, M.D., Peter Richardson, M.R.C.P., Ulrich Trechsel,
M.D., Albert Widmer, Dipl.Stat., Jean-Pierre Devogelaer, M.D., Jean-Marc
Kaufman, M.D., Ph.D., Philippe Jaeger, M.D., Jean-Jacques Body, M.D., Ph.D.,
Maria Luisa Brandi, M.D., Johann Broell, M.D., Raffaele Di Micco, M.D., Andrea
Riccardo Genazzani, M.D., Dieter Felsenberg, M.D., Joachim Happ, M.D., Michael
J. Hooper, F.R.A.C.P., Jochen Ittner, M.D., Georg Leb, M.D., Hans Mallmin,
M.D., Ph.D, Timothy Murray, M.D., Sergio Ortolani, M.D., Alessandro Rubinacci,
M.D., Maria Sääf, M.D., Ph.D., Goran Samsioe, M.D., Ph.D., Leon Verbruggen,
M.D., Ph.D., and Pierre J. Meunier, M.D.
ABSTRACT
Background Bisphosphonates
are effective agents for the management of osteoporosis. Their low
bioavailability and low potency necessitate frequent administration
on an empty stomach, which may reduce compliance. Gastrointestinal
intolerance limits maximal dosing. Although intermittent intravenous
treatments have been used, the optimal doses and dosing interval have
not been systematically explored.
Methods We studied
the effects of five regimens of zoledronic acid, the most potent
bisphosphonate, on bone turnover and density in 351 postmenopausal
women with low bone mineral density in a one-year, randomized, double-blind,
placebo-controlled trial. Women received placebo or intravenous
zoledronic acid in doses of 0.25 mg, 0.5 mg, or 1 mg at three-month
intervals. In addition, one group received a total annual dose of 4
mg as a single dose, and another received two doses of 2 mg each,
six months apart. Lumbar-spine bone mineral density was the primary
end point.
Results There were
similar increases in bone mineral density in all the zoledronic acid
groups to values for the spine that were 4.3 to 5.1 percent higher
than those in the placebo group (P<0.001) and values for the
femoral neck that were 3.1 to 3.5 percent higher than those in the
placebo group (P<0.001). Biochemical markers of bone resorption
were significantly suppressed throughout the study in all zoledronic
acid groups. Myalgia and pyrexia occurred more commonly in the
zoledronic acid groups, but treatment-related dropout rates were
similar to that in the placebo group.
Conclusions Zoledronic
acid infusions given at intervals of up to one year produce effects
on bone turnover and bone density as great as those achieved with
daily oral dosing with bisphosphonates with proven efficacy against
fractures, suggesting that an annual infusion of zoledronic acid
might be an effective treatment for postmenopausal osteoporosis.
Oral bisphosphonates are widely used for
treating osteoporosis and have been shown to increase bone mineral
density and decrease the rate of fracture.1,2
However, they do have limitations related to long-term compliance,
gastrointestinal intolerance, and poor and variable absorption from
the gastrointestinal tract. Intermittent intravenous administration
of bisphosphonates might address some of these problems and has been
shown to be effective in the treatment of malignant hypercalcemia
and Paget's disease and to reduce the rate of skeletal complications
in patients with breast carcinoma or multiple myeloma. Evidence
suggests that intravenous bisphosphonates increase bone mineral
density in patients with osteoporosis, but most relevant studies
have been small, unblinded, and short-term and have not
systematically examined the effects of the dose and dosing interval
on changes in bone mineral density and markers of bone turnover.3,4,5,6
Zoledronic acid is the most potent bisphosphonate that has been
studied in clinical trials to date.7 It
is superior to pamidronate in the treatment of cancer-related
hypercalcemia.8
Because it has high potency, only small doses are required for the
inhibition of bone resorption, and long dosing intervals may be
used. We undertook a phase 2 study to examine the effect of
intravenous zoledronic acid on bone density and bone turnover in
postmenopausal women with low bone density and to assess the effects
of varying the total dose administered and the dosing interval.
Methods
Study Subjects
A total of 351 postmenopausal women 45 to 80 years of age were
studied at 24 centers in 10 countries. In all the women, menopause had
occurred at least five years previously, either naturally or as the
result of bilateral oophorectomy. All women had a bone mineral
density at the lumbar spine (L1 to L4) that was at least 2.0 SD
below the mean value for young adults (a T score lower than –2) and
had no more than one vertebral fracture at screening. The date of
onset of menopause was defined as the date of oophorectomy when
applicable or as 12 months after the cessation of menses in women
over 50 years of age and 18 months after the cessation of menses in
women between 45 and 49 years of age. Major criteria for exclusion
included systemic estrogen treatment within the previous three
months, evidence of secondary osteoporosis, clinical or laboratory
evidence of hepatic or renal disease, disorders of the parathyroid
or thyroid glands, a serum 25-hydroxyvitamin D concentration of 15
ng per milliliter (37 nmol per liter) or less, a history of cancer,
previous treatment with bisphosphonates or fluoride, and current therapy
with any other drug known to affect the skeleton. The protocol was
approved by the ethics committee at each center, and all the women
gave written informed consent. Thirty-five women withdrew from the
study, most commonly for personal reasons (in the case of 15 women)
or because of adverse events (14 women). Thus, 316 women completed
the study.
Treatment
All women received a calcium supplement (1 g per day). At study
entry the women were randomly assigned to receive one of six treatment
regimens in a double-blind fashion. Three groups received zoledronic
acid by intravenous infusion every three months, one group at a dose
of 0.25 mg, one at a dose of 0.5 mg, and one at a dose of 1 mg. Two
other groups received a total dose of 4 mg of zoledronic acid — one
group receiving a single 4-mg infusion at the beginning of the trial
and the other group receiving two doses of 2 mg each, one at base
line and the other at six months. Thus, there were three groups that
received a total dose of 4 mg in one year. The sixth group received
only placebo (saline). To maintain blinding, all women received an
intravenous infusion of either zoledronic acid or placebo every three
months. All infusions were 20 ml in volume and were infused over a
period of five minutes. A dose of 4 mg given in this way produces a
mean (±SD) peak serum concentration of zoledronic acid of 393±100 ng
per milliliter. Infusions were prepared at each center by a
pharmacist who had no contact with the patients and were labeled
with the subject's study number and supplied to the study personnel.
Bone Density Measurement
The bone mineral density of the lumbar spine, the nondominant
proximal femur and forearm, and the total body were measured by
dual-energy x-ray absorptiometry at base line and at 6, 9, and 12
months with the use of Hologic QDR (Hologic, Waltham, Mass.) or
Lunar (Madison, Wis.) instruments. Data were converted to
Hologic-equivalent values by the method of Hui et al.9 A
central laboratory (Institut für Funktionsanalyse, Hamburg, Germany)
was responsible for the supervision of quality control for these
measurements and notified investigators if any patient had a
decrease in bone density of more than 5 percent from the base-line
values.
Markers of Bone Turnover
Measurement of biochemical markers was performed in a central
laboratory with the use of established methods. For serum bone-specific
alkaline phosphatase, the Tandem-MP Ostase assay was used (Hybritech,
Liege, Belgium). Serum osteocalcin was measured with the N-MID one-step
enzyme-linked immunosorbent assay (Osteometer, Herlev, Denmark).
Urinary type I collagen cross-linked N-telopeptide was measured with
the Osteomark assay (Ostex, Seattle). Serum type I collagen C-telopeptide
was measured with the CrossLaps assay (Osteometer).
Statistical Analysis
The necessary sample size was calculated as the number of patients
needed to detect a difference between the zoledronic acid groups and
the placebo group of at least 4 percent in the degree of change in
lumbar-spine bone mineral density from base line to 12 months.
Bonferroni's correction was used to adjust for multiple comparisons
in order to ensure an overall nominal significance level of 0.05.
Given a noncentral t distribution with a type I error of 0.025, a
power of 80 percent, a two-sided alternative, and a standard
deviation of 5.7 percent, we calculated that 40 patients were needed
in each treatment group in order to allow detection of a difference
of 4 percent. To allow for a possible 15 percent dropout rate, a
total sample size of 290 was selected.
All analyses were performed according to the intention-to-treat
principle with the use of all available data from all patients who
received study drug. Missing values were not imputed or replaced.
Analysis of covariance was performed (with the Proc Mixed procedure
of SAS software [SAS Institute, Cary, N.C.]) to estimate differences
between the treatment groups. The statistical fixed-effects model
considered center and treatment as main variables. In addition, the
base-line values, if measured, were used as covariates. The analyses
were repeated with the last observation carried forward and produced
essentially the same results (data not shown).
For the primary variable, adjustment for multiple comparisons
between placebo and the active doses of zoledronic acid was performed
at a one-sided alpha level of 0.025, according to the method of
Marcus et al.10
For secondary variables, pairwise comparisons were investigated in
the exploratory analysis (unadjusted for multiple comparisons). The
pairwise comparisons were tested at a two-sided level of
significance of 0.05. In addition to the P value for the comparisons
between treatment groups, estimates of the differences and
associated 95 percent confidence intervals were calculated.
The protocol was designed and developed by the sponsor and
submitted to the investigators for comments and amendments. The
final protocol was then accepted by the investigators and submitted
to the ethics review committees of their institutions for approval. Data
management and statistical analysis were performed by the sponsor.
Interpretation of the data and preparation of the manuscript were
performed by a publication committee that included three academic
researchers who were investigators in the trial (Drs. Reid, Brown,
and Burckhardt) and Dr. Trechsel, the author of the study protocol,
as a representative of the sponsor. These authors had full and
unfettered access to the data and take full responsibility for the
completeness and accuracy of the reported data. The study sponsor
placed no limits on statements made in the final paper.
Results
Study Subjects
The base-line characteristics of the women who participated in
the study are summarized in Table 1. All but
two women were white, and none had vertebral fractures at study
entry.
|
Bone Mineral Density
Mean bone-mineral-density values in the lumbar spine corresponded
to a T score of –2.9. All groups receiving zoledronic acid regimens
had a progressive increase in bone mineral density in the lumbar
spine throughout the 12-month study period, although the rate of
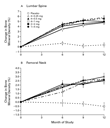
increase tended to slow in the second half of the study (Figure 1A).
Throughout the study, the values for lumbar-spine bone mineral
density achieved with all zoledronic acid regimens were
significantly higher than those in the placebo group (P<0.001), and
there were no significant differences among the zoledronic acid
groups. At 12 months, the mean lumbar-spine bone mineral density in
the groups receiving zoledronic acid was 4.3 to 5.1 percent higher
than the mean value in the placebo group, which remained stable. The
bone mineral density in the femoral neck also increased
progressively throughout the study period; all zoledronic acid
groups had similar increases to values that were significantly
higher than those in the placebo group (differences of 3.1 to 3.5
percent at 12 months, P<0.001) (Figure 1B). The
femoral-neck bone mineral density declined by 0.4 percent in the
placebo group.
|
Bone mineral density at the distal radius responded to zoledronic acid
treatment to a lesser extent, resulting in differences from the
placebo group of 0.8 to 1.6 percent at 12 months (data not shown);
in the placebo group, distal radial bone mineral density decreased
by 0.8 percent. All zoledronic acid regimens except the four doses
of 0.25 mg each resulted in distal radial bone mineral density that
was significantly greater than that in the placebo group (P![]() 0.05 for all comparisons).
The results for total-body bone mineral density were similar (data
not shown). At 12 months, the differences in total-body bone mineral
density between the zoledronic acid groups and the placebo group
ranged from 0.9 percent to 1.3 percent and were significant
(P<0.03 for all comparisons) for all regimens except the four
doses of 0.5 mg each.
0.05 for all comparisons).
The results for total-body bone mineral density were similar (data
not shown). At 12 months, the differences in total-body bone mineral
density between the zoledronic acid groups and the placebo group
ranged from 0.9 percent to 1.3 percent and were significant
(P<0.03 for all comparisons) for all regimens except the four
doses of 0.5 mg each.
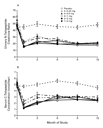
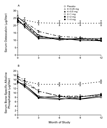
Markers of Bone Turnover
Markers of bone resorption reached a nadir at one month (median
decreases of 65 to 83 percent in serum C-telopeptide and 50 to
69 percent in the urinary N-telopeptide:creatinine ratio), whereas
there were no significant changes in the placebo group (Figure 2). The decrease
in markers of resorption tended to be dose-dependent, particularly
at three months — a pattern that is consistent with previous reports
that higher doses of bisphosphonates increase the duration of action
of the drug.11
We do not have full documentation of the immediate reductions in
bone resorption after each infusion, because most samples were
obtained only every three months. The suppression of resorption was
maintained at 12 months. At 12 months, the zoledronic acid regimens
were associated with decreases of 49 to 52 percent in serum
C-telopeptide (as compared with a decrease of 8 percent in the
placebo group) and decreases of 54 to 65 percent in the ratio of
urinary N-telopeptide to creatinine (as compared with an increase of
3 percent in the placebo group). All zoledronic acid groups had
values for these markers of resorption that were significantly
different from those in the placebo group (P<0.01 for all
comparisons), but there were no significant differences among the
zoledronic acid groups. Bone-specific alkaline phosphatase and
osteocalcin, which are serum markers of bone formation, showed
similar responses, but there was no sharp decrease apparent at one
month (Figure 3).
Again, suppression persisted at 12 months with all doses
(P<0.001).
|
|
Bone Biopsies
A 7.5-mm transiliac biopsy specimen was obtained from 43 women
and double-labeled with tetracycline. Of these specimens, 27 were
complete and suitable for histomorphometric analysis. The sections
were undecalcified and stained with Goldner's trichrome, except for
tetracycline measurements, which were made on unstained sections.
Women treated with zoledronic acid at any dose had significantly
lower proportions of mineralizing surfaces, rates of bone formation,
adjusted mineral apposition rates, and activation frequencies than
the women in the placebo group (differences of 71 percent to 84
percent, P<0.05); there were nonsignificant differences in the
proportion of eroded surface (39 percent lower than that in the
placebo group, P<0.06) and in eroded volume (48 percent lower,
P<0.07). No change was noted in cortical bone thickness or
porosity; cancellous bone volume; trabecular thickness, separation,
or number; wall width of trabecular bone packets; number of nodes
per volume of tissue; or osteoid maturation time. No dose effect was
found with respect to any of these factors. No evidence of
osteomalacia was found, either by qualitative assessment or on the
basis of such quantitative measures as osteoid thickness and volume
or the mineral apposition rate. No other qualitative abnormalities
were apparent.
Fractures
Spinal radiographs at base line and one year showed no vertebral
fractures during the study. No nonvertebral fractures occurred in
the group receiving four doses of 0.25 mg of zoledronic acid; two
nonvertebral fractures occurred in the group receiving four doses of
1 mg of zoledronic acid; and one nonvertebral fracture occurred in
each of the other groups.
Safety
Mean serum calcium concentrations in the zoledronic acid groups
declined significantly (P<0.05 for all comparisons), by approximately
0.08 mmol per liter, between base line and one month but were similar
to those in the placebo group from three months onward. Serum
phosphate concentrations in the zoledronic acid groups had decreased
by 0.06 to 0.12 mmol per liter at one month and generally remained
about 0.05 mmol per liter below those in the placebo group
throughout the study period, although they did not differ
significantly from those in the placebo group at one year. Intact
parathyroid hormone was measured in serum at base line and 12
months. There were no significant differences among the groups at
the 12-month follow-up, although the mean value was about 30 percent
higher than the base-line value in the women in the group receiving
four doses of 1 mg of zoledronic acid, possibly because sampling was
performed only three months after the last dose had been
administered in this group.
The rates of adverse events were similar in all the
active-treatment groups (Table 2).
However, treatment-related adverse events were significantly more
common in the zoledronic acid groups than in the placebo group
(rates of 45 to 67 percent vs. 27 percent; data not shown). In the zoledronic
acid groups, most adverse events were instances of musculoskeletal
pain, nausea, or fever, most of which were rated as mild. Most
occurred the first time the drug was administered. Five women
withdrew from the study because of drug-related adverse events, all
of which were reactions after the first infusion of zoledronic acid.
These withdrawals were not dose-related; two occurred in women who
were receiving the lowest dose and two in women receiving the
highest dose. There was no evidence of adverse effects on renal
function with any of these regimens. Overall, the proportions of
women who withdrew from the study because of adverse events were
similar in all groups. Symptoms at the infusion site were uncommon
in all groups (e.g., reported in no patients receiving a single 4-mg
dose of zoledronic acid and in two patients receiving placebo).
Iritis did not develop in any patients, and the occurrence of any
eye disorder was uncommon (e.g., reported in two patients receiving
a single 4-mg dose of zoledronic acid and in nine patients receiving
placebo).
|
Discussion
Intermittent intravenous administration of the potent
bisphosphonate zoledronic acid results in changes in biochemical
markers of bone turnover and in bone mineral density that are
similar to those observed with daily oral bisphosphonate therapy.
Thus, the reductions in markers at one year in the present study are
similar to those seen with 5 mg of risedronate per day,12
2.5 to 5 mg of ibandronate per day,13
and 10 mg of alendronate per day.14,15,16 Zoledronic
acid increases spinal bone mineral density at 12 months to 5 percent
above values found in patients receiving placebo — an increase
similar to that achieved with a daily 10-mg dose of alendronate (5
percent),17
a daily 5-mg dose of risedronate (3 percent),12 or
a daily 150-mg dose of pamidronate (5 percent).18
Intravenous zoledronic acid also produced results similar to those
of the oral regimens at the femoral neck (alendronate, 3 percent
increase in bone density; risedronate, 2 percent; pamidronate, 3
percent) and in the total body (alendronate, 1.5 percent increase;
pamidronate, 1 percent).
Our study assessed longer intervals between doses than have been
assessed by previous studies of intermittent bisphosphonate therapy.
Etidronate has been used for many years in two-week oral courses
administered at three-month intervals.19,20
There is also evidence that intravenous pamidronate3 or
ibandronate,4
given every three months, has beneficial effects on bone density in
women with postmenopausal osteoporosis. The disappointing data on
fractures from a recent study of intermittent ibandronate therapy (1
mg intravenously every three months)21
has been interpreted as indicating that a dosing interval of three
months is too long. However, this ibandronate regimen did not stably
suppress markers of bone resorption; a substantial maximal
suppression of C-telopeptide excretion (by 50 percent) was rapidly
offset, so that the level before the next dose was only 10 to 20
percent below that in the placebo group.4 As a
result, the changes in bone density (increases of 2.9 percent in the
spine at 12 months4
or to 4 percent higher than the spinal bone mineral density in
the placebo group at 3 years21)
were smaller than those found in our study; this effect is
consistent with the moderate effect of this dose of ibandronate on
the incidence of vertebral fracture (a 26 percent reduction at 3
years). Our data indicate that much longer dosing intervals are
compatible with efficacy (in terms of both suppression of bone
turnover and increase in bone density) if the dose of bisphosphonate
is sufficiently large. Indeed, the present study does not establish
a maximal dosing interval, since turnover remained suppressed at 12
months. Thus, it is possible that a longer interval between doses
could be effective, particularly if larger doses of zoledronic acid
were used.
How a single infusion of zoledronic acid suppresses bone turnover
for so long remains to be determined. Prolonged suppression is
not the result of the persistence of the drug in the circulation, given
that by 24 hours after administration, drug levels are less than 1
percent of the postadministration peak and 40 percent of the dose
has been excreted in the urine. The balance of the dose is
presumably bound to bone and is slowly released back into the
circulation, giving rise to a 167-hour terminal half-life in plasma.
It has been thought that bisphosphonates are located exclusively on
osteoclastic surfaces22
and that short-term exposure inhibits activity in a single
generation of basic multicellular units in bone. The life span of
the basic multicellular unit (about three months) then determines the
duration of action of the drug. However, evidence suggests that
bisphosphonates are also deposited on osteoblastic and resting bone
surfaces and remain there for the long term.23 The
existence of such deposits would provide a possible explanation for
our results, since residue from a single dose could interfere with
the future development of basic multicellular units at these
surfaces. It is also possible that direct effects on existing basic
multicellular units and osteocytes24,25
result in reduced formation of succeeding basic multicellular units.
Zoledronic acid was generally well tolerated, and the rate of
retention of subjects in the study was high. The adverse events that
were more common in women receiving zoledronic acid are those that
have occurred previously in patients receiving intravenous aminobisphosphonates
and are transient. Infrequent doses may increase tolerance of these
side effects.
The inclusion of a placebo group in this study permits
quantification of the size of the therapeutic effect and facilitates
comparison of the present data with those from other studies. We
believe this use of a placebo is ethical, since the bone density
used as a criterion for entry (a T score of less than –2) is higher
than that required at the participating centers for a diagnosis of
osteoporosis and would certainly not be considered to be a threshold
for therapeutic intervention at these centers. Thus, the study was
conducted in a low-risk population — a characterization supported by
the fact that no spinal fractures occurred during the study period.
Only one sixth of these low-risk subjects received placebo, and they
received it for a maximum of 12 months, after which all women
received active therapy.
Osteoporosis has been regarded as requiring daily therapy, and
maintaining compliance with daily regimens for a predominantly asymptomatic
condition has been a major problem.26,27 Administration
of treatment at intervals of 6 to 12 months or more is likely to
be much more acceptable to patients and could reduce costs. A
greater proportion of the at-risk population might take advantage of
prophylaxis against osteoporosis if an intermittent regimen were
used, and the rate of fractures might therefore decrease. However,
studies that demonstrate an effect on the rate of fractures are
needed before any recommendation can be made.
Supported
by a grant from Novartis Pharma.
We are indebted to Esther Hagin of Novartis Pharma, Basel,
Switzerland, for her expert work in conducting the trial.
Source Information
From the Department of Medicine, University of Auckland, Auckland,
New Zealand (I.R.R.); the Centre de Recherche du Centre Hospitalier del
Université Laval, Quebec, Que., Canada (J.P.B.); the Centre Hospitalier
Universitaire Vaudois, Lausanne, Switzerland (P.B.); Novartis Pharmaceuticals,
East Hanover, N.J. (Z.H., P.R.); Novartis Pharma, Basel, Switzerland (U.T.,
A.W.); the Department of Rheumatology, Université Catholique de Louvain, St.
Luc, Brussels, Belgium (J.-P.D.); the Department of Endocrinology, University
Hospital of Ghent, Ghent, Belgium (J.-M.K.); the Medizinische
Universitätspoliklinik, Inselspital Bern, Bern, Switzerland (P.J.); the
Endocrinology and Supportive Care Clinic, Institut J. Bordet, Free University
of Brussels, Brussels, Belgium (J.-J.B.); and the Department of Rheumatology
and Bone Diseases, Hôpital Edouard Herriot, Lyons, France (P.J.M.).
Other authors were Maria Luisa Brandi, M.D., Unità di Endocrinologia, Ospedale
di Careggi, Florence, Italy; Johann Broell, M.D., Medizinische Abteilung mit
Rheumatologie und Osteologie, Kaiser-Franz-Josef-Spital der Stadt Wien, Vienna,
Austria; Raffaele Di Micco, M.D., Centro di Fisiopatologia della Menopausa,
Ospedale Maggiore La Maternità, Bologna, Italy; Andrea Riccardo Genazzani,
M.D., Ospedali Riuniti S. Chiara, Pisa, Italy; Dieter Felsenberg, M.D.,
Universitätsklinikum Benjamin Franklin, Strahlenklinik und Poliklinik, Berlin,
Germany; Joachim Happ, M.D., Frankfurt, Germany; Michael J. Hooper, F.R.A.C.P.,
Department of Endocrinology, Concord Hospital, Concord, N.S.W., Australia;
Jochen Ittner, M.D., Augsburg, Germany; Georg Leb, M.D., Klinische Abteilung
für Endokrinologie und Nuklearmedizin, Medizinische Universitätsklinik, Graz,
Austria; Hans Mallmin, M.D., Ph.D., Medicinkliniken, Akademisca Sjukhuset,
Uppsala, Sweden; Timothy Murray, M.D., Metabolic Bone Clinic, St. Michael's
Hospital, Toronto; Sergio Ortolani, M.D., Centro Studi Metabolismo Osseo,
Istituto Auxologico Italiano, Milan, Italy; Alessandro Rubinacci, M.D., Unità
Metabolica dell'Osso, Ospedale S. Raffaele, Milan, Italy; Maria Sääf, M.D.,
Ph.D., Endokrinologiska Kliniken, Karolinska Sjukhuset, Stockholm, Sweden;
Goran Samsioe, M.D., Ph.D., Kvinnokliniken, Universitetskliniken, Lund, Sweden;
and Leon Verbruggen, M.D., Ph.D., Department of Rheumatology, Academic
Hospital, Brussels Free University, Brussels, Belgium.
Address reprint requests to Professor Reid at the Department of
Medicine, University of Auckland, Private Bag 92019, Auckland, New Zealand, or
at [log in to unmask].
References
- Black
DM, Cummings SR, Karpf DB, et al. Randomised trial of effect of
alendronate on risk of fracture in women with existing vertebral
fractures. Lancet 1996;348:1535-1541.[Medline]
- McClung
MR, Geusens P, Miller PD, et al. Effect of risedronate on the risk of hip
fracture in elderly women. N Engl J Med 2001;344:333-340.[Abstract/Full
Text]
- Thiebaud
D, Burckhardt P, Melchior J, et al. Two years' effectiveness of
intravenous pamidronate (APD) versus oral fluoride for osteoporosis
occurring in the postmenopause. Osteoporos Int 1994;4:76-83.[Medline]
- Thiebaud
D, Burckhardt P, Kriegbaum H, et al. Three monthly intravenous injections
of ibandronate in the treatment of postmenopausal osteoporosis. Am J Med
1997;103:298-307.[Medline]
- Khan SA,
Kanis JA, Vasikaran S, et al. Elimination and biochemical responses to
intravenous alendronate in postmenopausal osteoporosis. J Bone Miner Res
1997;12:1700-1707.[Medline]
- Passeri
M, Baroni MC, Pedrazzoni M, et al. Intermittent treatment with intravenous
4-amino-1-hydroxybutylidene-1,1-bisphosphonate (AHBuBP) in the therapy of
postmenopausal osteoporosis. Bone Miner 1991;15:237-247.[Medline]
- Body JJ,
Lortholary A, Romieu G, Vigneron AM, Ford J. A dose-finding study of
zoledronate in hypercalcemic cancer patients. J Bone Miner Res
1999;14:1557-1561.[Medline]
- Major P,
Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in
the treatment of hypercalcemia of malignancy: a pooled analysis of two
randomized, controlled clinical trials. J Clin Oncol 2001;19:558-567.[Abstract/Full
Text]
- Hui SL,
Gao SJ, Zhou XH, et al. Universal standardization of bone density
measurements: a method with optimal properties for calibration among
several instruments. J Bone Miner Res 1997;12:1463-1470.[Medline]
- Marcus
R, Peritz E, Gabriel KR. On closed testing procedures with special
reference to ordered analysis of variance. Biometrika 1976;63:655-660.
- Thiébaud
D, Husi B, Jacquet AF, Burckhardt P. Effects of ibandronate i.v. bolus
injection in healthy men and postmenopausal women. J Bone Miner Res
1997;12:Suppl 1:S343-S343.abstract
- Harris
ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on
vertebral and nonvertebral fractures in women with postmenopausal osteoporosis:
a randomized controlled trial. JAMA 1999;282:1344-1352.[Medline]
- Ravn P,
Clemmesen B, Riis BJ, Christiansen C. The effect on bone mass and bone
markers of different doses of ibandronate: a new bisphosphonate for
prevention and treatment of postmenopausal osteoporosis: a 1-year,
randomized, double-blind, placebo-controlled dose-finding study. Bone
1996;19:527-533.[Medline]
- Garnero
P, Shih WCJ, Gineyts E, Karpf DB, Delmas PD. Comparison of new biochemical
markers of bone turnover in late postmenopausal osteoporotic women in
response to alendronate treatment. J Clin Endocrinol Metab
1994;79:1693-1700.[Abstract]
- Devogelaer
JP, Broll H, Correa-Rotter R, et al. Oral alendronate induces progressive
increases in bone mass of the spine, hip, and total body over 3 years in
postmenopausal women with osteoporosis. Bone 1996;18:141-150. [Erratum,
Bone 1996;19:78.][Medline]
- Chesnut
CH III, McClung MR, Ensrud KE, et al. Alendronate treatment of the
postmenopausal osteoporotic woman: effect of multiple dosages on bone mass
and bone remodeling. Am J Med 1995;99:144-152.[Medline]
- Liberman
UA, Weiss SR, Bröll J, et al. Effect of oral alendronate on bone mineral
density and the incidence of fractures in postmenopausal osteoporosis. N
Engl J Med 1995;333:1437-1443.[Abstract/Full
Text]
- Reid IR,
Wattie DJ, Evans MC, Gamble GD, Stapleton JP, Cornish J. Continuous
therapy with pamidronate, a potent bisphosphonate, in postmenopausal
osteoporosis. J Clin Endocrinol Metab 1994;79:1595-1599.[Abstract]
- Storm T,
Thamsborg G, Steiniche T, Genant HK, Sørensen OH. Effect of intermittent
cyclical etidronate therapy on bone mass and fracture rate in women with
postmenopausal osteoporosis. N Engl J Med 1990;322:1265-1271.[Abstract]
- Watts
NB, Harris ST, Genant HK, et al. Intermittent cyclical etidronate
treatment of postmenopausal osteoporosis. N Engl J Med 1990;323:73-79.[Abstract]
- Recker
RR, Stakkestad JA, Felsenberg D, et al. A new treatment paradigm:
quarterly injections of ibandronate reduce the risk of fractures in women
with postmenopausal osteoporosis (PMO): results of a 3-year trial.
Osteoporos Int 2000;11:Suppl 2:S209-S209.abstract
- Sato M,
Grasser W, Endo N, et al. Bisphosphonate action: alendronate localization
in rat bone and effects on osteoclast ultrastructure. J Clin Invest
1991;88:2095-2105.[Medline]
- Masarachia
P, Weinreb M, Balena R, Rodan GA. Comparison of the distribution of 3H-alendronate
and 3H-etidronate in rat and mouse bones. Bone 1996;19:281-290.[Medline]
- Reinholz
GG, Getz B, Pederson L, et al. Bisphosphonates directly regulate cell
proliferation, differentiation, and gene expression in human osteoblasts.
Cancer Res 2000;60:6001-6007.[Abstract/Full
Text]
- Plotkin
LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T.
Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and
calcitonin. J Clin Invest 1999;104:1363-1374.[Abstract/Full
Text]
- Faulkner
DL, Young C, Hutchins D, McCollam JS. Patient noncompliance with hormone
replacement therapy: a nationwide estimate using a large prescription
claims database. Menopause 1998;5:226-229.[Medline]
- Christensen
PM, Kristiansen IS, Brøsen K, Brixen K, Abrahamsen B, Andersen M.
Compliance to bisphosphonates -- a population-based study using a drug
prescription database. Bone 2000;27:Suppl:21S-21S.abstract
Edward E.
Rylander, M.D.
Diplomat American
Board of Family Practice.
Diplomat American
Board of Palliative Medicine.