|
|

|
|
Widespread Coronary Inflammation in Unstable
Angina
Antonino Buffon, M.D., Luigi M. Biasucci, M.D., Giovanna
Liuzzo, M.D., Giuseppe D'Onofrio, M.D., Filippo Crea, M.D., and Attilio Maseri,
M.D.
ABSTRACT
Background
Inflammation within vulnerable coronary plaques may cause unstable
angina by promoting rupture and erosion. In unstable angina,
activated leukocytes may be found in peripheral and coronary-sinus
blood, but it is unclear whether they are selectively activated in
the vascular bed of the culprit stenosis.
Methods We measured
the content neutrophil myeloperoxidase content in the cardiac and
femoral circulations in five groups of patients: two groups with
unstable angina and stenosis in either the left anterior descending
coronary artery (24 patients) or the right coronary artery (9
patients); 13 with chronic stable angina; 13 with variant angina and
recurrent ischemia; and 6 controls. Blood samples were taken from
the aorta, the femoral vein, and the great cardiac vein, which
selectively drains blood from the left but not the right coronary
artery.
Results The
neutrophil myeloperoxidase content of aortic blood was similar in
both groups of patients with unstable angina (–3.9 and –5.5, with
negative values representing depletion of the enzyme due to
neutrophil activation) and significantly lower than in the other
three groups (P<0.05). Independently of the site of the stenosis,
the neutrophil myeloperoxidase content in blood from the great
cardiac vein was significantly decreased in both groups of patients
with unstable angina (–6.4 in those with a left coronary lesion and
–6.6 in those with a right coronary lesion), but not in patients
with stable angina and multiple stenoses, patients with variant angina
and recurrent ischemia, or controls. There was also a significant
transcoronary reduction in myeloperoxidase content in both groups with
unstable angina.
Conclusions The
widespread activation of neutrophils across the coronary vascular
bed in patients with unstable angina, regardless of the location of
the culprit stenosis, challenges the concept of a single vulnerable
plaque in unstable coronary syndromes.
The hypothesis that inflammation of a
vulnerable plaque is responsible for the development of acute
coronary syndromes1,2,3,4,5 is
stimulating a variety of techniques for the detection and stabilization
of vulnerable plaques.6,7,8,9,10
Yet, it is unclear whether the inflammatory process is confined to a
single vulnerable plaque or whether it is more widespread in the
coronary vasculature.
The possibility of widespread inflammation of the coronary
arterial bed is suggested by the recent report of multiple complex
coronary plaques in patients with acute myocardial infarction11
and by previous postmortem findings of multiple fresh thrombi in
patients with unstable angina12
and of multiple fissured, thrombosed plaques.13,14 A
widespread acute inflammatory process in the coronary arterial bed
would have important implications for a clearer understanding of the
pathogenesis, and eventually for the treatment and prevention, of
acute coronary syndromes. By "widespread," we mean
involvement of more than one major coronary artery. By measuring
leukocyte expression of CD11b and CD18 in aortic and coronary-sinus
blood, Mazzone et al.15
and de Servi et al.16
demonstrated a transcoronary inflammatory activation of monocytes
and neutrophils in patients with unstable angina. Such activation
was not detectable in aortic blood. Unfortunately, these authors did
not assess the correlation between activation and the location of
the culprit coronary stenosis responsible for the angina.15,16
Marked activation of neutrophils was also detected in the peripheral
blood of patients with unstable angina, but not in those with stable
angina or in controls. Activation was detected by measuring the
neutrophil myeloperoxidase content, which is an index of more
advanced inflammatory activation than that identified by measuring
CD11b and CD18 expression.17,18
We ascertained whether the activation of neutrophils, presumably
due to inflammation, in patients with unstable angina was confined to
the vascular bed perfused by the vessel with the culprit coronary
stenosis, or whether it also involved the vascular bed of
angiographically normal or nearly normal arteries. We selected
patients with coronary stenoses of either the left anterior
descending or the right coronary artery. We simultaneously measured
the neutrophil myeloperoxidase content in blood from the aorta, the
femoral vein, and the great cardiac vein, which selectively drains
blood from the left anterior descending coronary artery but not the
right coronary artery.19
Patients with stable angina and stenosis of the left anterior
descending coronary artery, patients with variant angina and
recurrent ischemia of the left anterior descending coronary artery,
and patients without coronary disease (controls) were also studied.
Methods
Patients
We studied a total of 65 patients, divided into five groups. Two
of the groups consisted of the 33 patients who had Braunwald class
IIIB unstable angina. Coronary angiography showed that the coronary
stenosis responsible for the angina (the culprit stenosis) was in
the left anterior descending coronary artery in 24 of these patients
(the first group), and in the right coronary artery in the other 9
patients (the second group). The remaining three groups were made up
of 13 patients with chronic stable angina and stenosis in the left
anterior descending coronary artery; 13 patients with active variant
angina and recurrent spasm in the left anterior descending coronary
artery, which was documented by testing with ergonovine; and 6
control patients with mild mitral stenosis, atrial septal defect, or
supraventricular tachycardia and a normal coronary angiogram.
Patients with a recent myocardial infarction (within three
months), prior coronary interventions, an occluded coronary vessel,
a culprit coronary stenosis in the circumflex branch, or
intercurrent infective or inflammatory disorders were excluded from
the study. No patients were taking antiinflammatory agents other
than aspirin (up to 100 mg daily).
The protocol was approved by the ethics committee of the Catholic
University of Rome, and all patients gave written informed consent.
Protocol
Serum levels of C-reactive protein were measured on admission
and used as a marker of systemic inflammation. Cardiac catheterization
was performed within a mean (±SD) of 2±1 days. Before the
injection of a contrast agent, all patients underwent sampling of
blood from the right femoral vein and simultaneous sampling of blood
from the aorta and great cardiac vein for the measurement of
neutrophil myeloperoxidase. In both groups of patients with unstable
angina, in order to demonstrate that the great cardiac vein
selectively drained blood from the left anterior descending but not
from the right coronary artery, the blood oxygen saturation in the
great cardiac vein was determined before and after the injection of
1.0 mg of isosorbide dinitrate into the left anterior descending or
the right coronary artery, according to the location of the culprit
stenosis. The venous–arterial differences in neutrophil and
leukocyte counts through the coronary and peripheral circulations
were also determined.
The myeloperoxidase content was determined by using a hematologic
analyzer (Bayer H*1), which measures the differential leukocyte count
as well as the cell count by automated flow cytochemistry, as
previously described.17
The H*1 computer software calculates a myeloperoxidase index of the
mean myeloperoxidase content in the neutrophil population. In
healthy subjects, this index is close to 0. Positive values
characterize neutrophils rich in myeloperoxidase, and negative
values characterize neutrophils depleted of myeloperoxidase as a
consequence of their activation. A lower myeloperoxidase index in
blood from the great cardiac vein or the femoral vein, as compared
with the aorta, was taken as an index of neutrophil activation
through the coronary or femoral vascular bed. C-reactive protein
levels were measured by a high-sensitivity, latex-enhanced immunonephelometric
assay (Dade Behring BN II analyzer).20
The working range of the assay was 0.175 to 1100 mg per liter, and
the coefficient of variation was less than 5 percent.
Statistical Analysis
Because the myeloperoxidase index did not have a normal
distribution, nonparametric tests were used: the Mann–Whitney test
and the Kruskal–Wallis test with multiple-comparison procedures
(Dunn's method) for comparisons between groups, and the Friedman test
and the Wilcoxon test with the Bonferroni correction for comparisons
within groups. Correlations were determined with use of Spearman's
rank-correlation coefficient.21
The leukocyte and neutrophil counts had a normal distribution and
were evaluated by analysis of variance for repeated measures with
the Bonferroni correction. Chi-square statistics were used for
categorical variables. A P value of less than 0.05 (two-tailed) was
considered to indicate statistical significance. Data are reported
as medians and ranges or as means ±SD, as appropriate.
Results
The demographic, clinical, and angiographic characteristics of
the patients are reported in Table 1 and Table 2. Anginal
symptoms before coronary angiography were similar in patients who
had unstable angina with a left coronary lesion, those who had
unstable angina with a right coronary lesion, and those who had
variant angina (Table
1).
|
|
The blood oxygen saturation in the great cardiac vein markedly increased
after injection of isosorbide dinitrate (1 mg) into the left
anterior descending coronary artery of patients who had unstable
angina with a left coronary lesion, but not after injection into the
right coronary artery of patients who had unstable angina with a
right coronary lesion (P=0.001 by two-way analysis of variance). The
median change in the blood oxygen saturation as a result of the
isosorbide dinitrate injection differed significantly between the
two groups (52.4 percent vs. 12.2 percent, P=0.04), indicating that
positioning the catheter in the great cardiac vein allowed for
selective sampling of the blood draining from the vascular bed of
the left anterior descending coronary artery. The leukocyte and
neutrophil counts in the aorta, great cardiac vein, and femoral vein
were similar; no differences were observed among groups (Table 1). Among
patients who had unstable angina with a right coronary lesion, the
territory of the left anterior descending coronary artery had no
wall irregularities in three patients, wall irregularities alone
in three patients, and stenosis of 30 to 50 percent of the luminal diameter
in the remaining three patients. Therefore, the atherosclerotic involvement
was much smaller than that observed in patients who had unstable
angina with a left coronary lesion and those who had chronic stable
angina (Table 2).22
Neutrophil Activation in the Systemic
Circulation
The median aortic myeloperoxidase indexes did not differ
significantly between patients who had unstable angina with a left
coronary lesion (–3.9) and those who had unstable angina with a
right coronary lesion (–5.5, P=0.21), but they were significantly lower
than those observed in patients with stable angina (+0.1), patients
with variant angina (+0.1), and controls (–0.8) (P<0.05 for all
comparisons). The ranges for all values are reported in Table 1 and
illustrated in Figure
1.
|
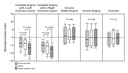
Neutrophil Activation through the Coronary and Femoral Circulations
In patients who had unstable angina with either a left or a right
coronary lesion, a significant transcoronary decrease in the
neutrophil myeloperoxidase index was observed. The median values in
blood from the aorta and the great cardiac vein were –3.9 and –6.4,
respectively, for those with a left-coronary-artery lesion
(P<0.001) and –5.5 and –6.6 for those with a
right-coronary-artery lesion (P=0.003). Conversely, no statistically
significant transcoronary decrease in neutrophil myeloperoxidase
content was observed in any of the other three groups; the
myeloperoxidase values in blood from the great cardiac vein were
+0.6 in patients with stable angina, –0.4 in those with variant
angina, and –0.2 in controls (P<0.01 for all the comparisons of
patients with stable angina, patients with variant angina, and
controls with both patients with unstable angina with a left
coronary lesion and those with unstable angina with a right coronary
lesion) (Table 1
and Figure 1).
No significant differences between the neutrophil myeloperoxidase
contents of aortic and femoral venous blood were observed in any of
the five groups (Table 1 and Figure 1).
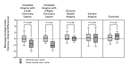
The change in neutrophil myeloperoxidase content across the coronary
circulation was significantly greater in both patients with unstable
angina with a left coronary lesion and those with unstable angina
with a right coronary lesion than in those with stable angina, those
with variant angina, and controls (Table 1 and
Figure 2). The
change in neutrophil myeloperoxidase content across the coronary
circulation was significantly greater than the difference in
neutrophil myeloperoxidase content between aortic and femoral venous
blood in both patients with unstable angina with a left coronary
lesion and those with unstable angina with a right coronary lesion,
but not in any of the other three groups (Table 1 and Figure 2).
|
Correlation between Levels of C-Reactive Protein and Myeloperoxidase
The median plasma levels of C-reactive protein were similar in
patients with unstable angina with a left coronary lesion (6.5 mg
per liter) and those with unstable angina with a right coronary
lesion (4.5 mg per liter, P=0.76) and were significantly higher than
the levels in patients with stable angina (2.1 mg per liter),
patients with variant angina (1.8 mg per liter), and controls (1.2
mg per liter; P<0.01 for all comparisons) (Table 1).
Overall, in the five groups, a significant correlation was found
between systemic levels of C-reactive protein and the aortic neutrophil
myeloperoxidase content (r=–0.45, P=0.03), as well as between
systemic levels of C-reactive protein and the neutrophil
myeloperoxidase content in blood from the great cardiac vein
(r=–0.41, P=0.01) (Figure
3).
|
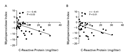
Discussion
Our findings confirm previous reports that in patients with unstable
angina, leukocytes become activated as they traverse the coronary
vascular bed,15,16
and that such activation may be detectable systematically.17,23,24 In
addition, we found no significant increase in neutrophil activation
in the great cardiac vein in controls, in patients with stable
angina and documented left anterior descending coronary stenosis, or
in patients with active variant angina and recurrent ischemia in
the territory of the left anterior descending coronary artery. Moreover,
there was no detectable increase in neutrophil activation through
the femoral circulation in any of the five groups studied.
In patients with unstable angina, transcoronary neutrophil
activation was not confined to the vascular bed perfused by the
artery in which the culprit stenosis was located and thus subjected
to recurrent ischemia. In fact, neutrophil activation occurred to
a similar extent in patients in whom the left anterior descending coronary
artery was not the site of the culprit stenosis. Patients with
unstable angina and a culprit lesion in the right coronary artery
had only minimal atherosclerotic involvement of the left anterior
descending coronary artery, which was angiographically normal in
three patients, had only luminal irregularities in three patients,
and had stenosis of less than 50 percent of the diameter in three
patients.
In animal models, neutrophil activation has been observed after
15 minutes of coronary occlusion–reperfusion.25
However, our findings cannot be explained simply on the basis of an ischemia–reperfusion
mechanism, in view of the fact that transcardiac neutrophil activation
was not observed in patients with active variant angina, spasm of
the left anterior descending artery, and a total ischemic burden
similar to that of patients with unstable angina.
In patients with unstable angina, inflammatory-cell infiltrates
are commonly found in most atherosclerotic plaques at postmortem examination1 and
in endarterectomy specimens.2,26
Multiple fissured, thrombosed coronary plaques seem to be a common
finding in acute coronary syndromes. Falk et al. reported 103
fissured, thrombosed plaques in 47 patients,13
and Davies and Thomas reported 111 fissured, thrombosed plaques in
76 patients.14
Neither of these reports discussed the possible clinical
significance of the simultaneous rupture of multiple plaques.
Multiple plaques with inflammatory-cell infiltrates and with a high
content of proinflammatory cytokines were reported by Arbustini et
al.12
Finally, multiple complex lesions were reported by Goldstein et al.11
The possibility that multiple plaque fissures and thrombi develop
simultaneously at different sites merely as a result of mechanical
stress seems rather unlikely. It would appear more reasonable to
speculate that a multifocal or widespread inflammatory activation of
the endothelium could change the characteristics of the interface
between the blood and the vessel walls from anticoagulant and vasodilative
to prothrombotic and vasoconstrictive, while at the same time
activating the metalloproteases and collagenases responsible for
endothelial-cell detachment and lysis of the plaque capsule at the
sites where it is weakest.
Whether neutrophils become activated by interacting with the surface
of sparse inflamed plaques or as a result of more widespread contact
with a diffusely inflamed coronary endothelium is not known. De
Servi et al. detected no activation of monocytes and neutrophils
across the culprit coronary stenosis in patients with unstable
angina.16
Conversely, the possibility of widespread coronary inflammation is
suggested by the reports of alterations in coronary flow27,28
and [18F]deoxyglucose uptake29 in
myocardial territories perfused by arteries without stenosis or
culprit lesions in patients with recent infarctions and in those
with unstable angina. Finally, in 10 percent of patients with
unstable angina, inflammatory red streaks were observed along
nonstenosed coronary arteries at the time of bypass surgery.30
The reported prevalence of systemically detectable inflammatory
markers in acute coronary syndromes varies. Serum levels of C-reactive
protein and of proinflammatory cytokines such as interleukin-6 are
elevated in about 70 percent of patients with severe unstable angina
on admission,31,32 in
50 percent of such patients at discharge, and in 45 percent of such
patients at six months of follow-up.20
These increased levels are associated with recurrent instability and
acute infarction. Accordingly, elevated levels of C-reactive protein
and interleukin-6 are found before the appearance of markers of
myocardial necrosis in nearly all patients in whom infarction is
preceded by unstable angina, but in less than 50 percent of patients
with myocardial infarction not preceded by unstable angina.31,33
Therefore, the triggers of coronary thrombosis and vasoconstriction
are not necessarily the same in all patients with acute coronary
syndromes.
The activation of neutrophils as they traverse the coronary circulation
of patients with unstable angina is a marker of a widespread
inflammatory process occurring in the coronary vasculature. When the
intensity of the inflammatory stimuli varies, such a process may
lead to waxing and waning of thrombosis and vasoconstriction. The
possibility of widespread coronary inflammation has important
implications for research and therapy. It challenges the widely
accepted hypothesis that a single vulnerable plaque is responsible
for the development of coronary instability — a hypothesis that is
currently stimulating the development of techniques for the
detection and stabilization of such plaques.
Supported by
grants from the National Research Council, Rome (94.00518.PF41), the
European Community (PL951505), and the Fondazione Internazionale di
Ricerca per il Cuore Onlus, Rome.
Source Information
From the Institute of Cardiology (A.B., L.M.B., G.L., F.C.) and
the Institute of Hematology (G.D.), Catholic University, Rome; and the
Cardiothoracic and Vascular Department, University Vita e Salute, Milan, Italy
(A.M.).
Address reprint requests to Dr. Maseri at the Cardiothoracic and
Vascular Department, University Vita e Salute, San Raffaele Hospital, Via
Olgettina 60, 20132 Milan, Italy, or at [log in to unmask].
References
- van
der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or
erosion of thrombosed coronary atherosclerotic plaques is characterized by
an inflammatory process irrespective of the dominant plaque morphology.
Circulation 1994;89:36-44.[Abstract]
- Moreno
PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage
infiltration in acute coronary syndromes: implications for plaque rupture.
Circulation 1994;90:775-778.[Abstract]
- Libby P.
Molecular bases of the acute coronary syndromes. Circulation
1995;91:2844-2850.[Full
Text]
- Shah PK,
Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce
collagen breakdown in fibrous caps of atherosclerotic plaques: potential
role of matrix-degrading metalloproteinases and implications for plaque
rupture. Circulation 1995;92:1565-1569.[ISI][Medline]
- Libby P.
Current concepts of the pathogenesis of the acute coronary syndromes.
Circulation 2001;104:365-372.[Full
Text]
- Casscells
W, Hathorn B, David M, et al. Thermal detection of cellular infiltrates in
living atherosclerotic plaques: possible implications for plaque rupture
and thrombosis. Lancet 1996;347:1447-1451.[ISI][Medline]
- Stefanadis
C, Diamantopoulos L, Vlachopoulos C, et al. Thermal heterogeneity within
human atherosclerotic coronary arteries detected in vivo: a new method of
detection by application of a special thermography catheter. Circulation
1999;99:1965-1971.[Abstract/Full
Text]
- Fayad
ZA, Fuster V, Fallon JT, et al. Noninvasive in vivo human coronary artery
lumen and wall imaging using black-blood magnetic resonance imaging.
Circulation 2000;102:506-510.[Abstract/Full
Text]
- Libby P.
Coronary artery injury and the biology of atherosclerosis: inflammation,
thrombosis, and stabilization. Am J Cardiol 2000;86:Suppl 2:3-8.[Medline]
- Rabbani
R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization.
Cardiovasc Res 1999;41:402-417.[ISI][Medline]
- Goldstein
JA, Demetriou D, Grines CL, Pica M, Shoukfeh M, O'Neill WW. Multiple
complex coronary plaques in patients with acute myocardial infarction. N
Engl J Med 2000;343:915-922.[Abstract/Full
Text]
- Arbustini
E, Grasso M, Diegoli M, et al. Coronary atherosclerotic plaques with and
without thrombus in ischemic heart syndromes: a morphologic, immunohistochemical,
and biochemical study. Am J Cardiol 1991;68:Suppl:36B-50B.[Medline]
- Falk E,
Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995;92:657-671.[Full
Text]
- Davies
MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden
cardiac ischemic death. N Engl J Med 1984;310:1137-1140.[Abstract]
- Mazzone
A, De Servi S, Ricevuti G, et al. Increased expression of neutrophil and
monocyte adhesion molecules in unstable coronary artery disease.
Circulation 1993;88:358-363.[Abstract]
- de Servi
S, Mazzone A, Ricevuti G, et al. Expression of neutrophil and monocyte
CD11B/CD18 adhesion molecules at different sites of the coronary tree in
unstable angina pectoris. Am J Cardiol 1996;78:564-568.[ISI][Medline]
- Biasucci
LM, D'Onofrio G, Liuzzo G, et al. Intracellular neutrophil myeloperoxidase
is reduced in unstable angina and myocardial infarction, but its reduction
is not related to ischemia. J Am Coll Cardiol 1996;27:611-616.[ISI][Medline]
- Hazen
SL, d'Avignon A, Anderson MM, Hsu FF, Heinecke JW. Human neutrophils
employ the myeloperoxidase-hydrogen peroxide-chloride system to oxidize
alpha-amino acids to a family of reactive aldehydes: mechanistic studies
identifying labile intermediates along the reaction pathway. J Biol Chem
1998;273:4997-5005.[Abstract/Full
Text]
- Ganz W,
Tamura K, Marcus HS, Donoso R, Yoshida S, Swan HJ. Measurement of coronary
sinus blood flow by continuous thermodilution in man. Circulation
1971;44:181-195.[ISI][Medline]
- Biasucci
LM, Liuzzo G, Grillo RL, et al. Elevated levels of C-reactive protein at
discharge in patients with unstable angina predict recurrent instability.
Circulation 1999;99:855-860.[Abstract/Full
Text]
- Alternatives
to analysis of variance and the t test based on ranks. In: Glantz SA.
Primer of biostatistics. 2nd ed. New York: McGraw-Hill, 1987:287-330.
- Bogaty
P, Brecker SJ, White SE, et al. Comparison of coronary angiographic
findings in acute and chronic first presentation of ischemic heart
disease. Circulation 1993;87:1938-1946.[Abstract]
- Mehta J,
Dinerman J, Mehta P, et al. Neutrophil function in ischemic heart disease.
Circulation 1989;79:549-556.[Abstract]
- Dinerman
JL, Mehta JL, Saldeen TG, et al. Increased neutrophil elastase release in
unstable angina pectoris and acute myocardial infarction. J Am Coll
Cardiol 1990;15:1559-1563.[ISI][Medline]
- Jordan
JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial
ischemia-reperfusion injury. Cardiovasc Res 1999;43:860-878.[ISI][Medline]
- DiSciascio
G, Cowley MJ, Goudreau E, Vetrovec GW, Johnson DE. Histopathologic
correlates of unstable ischemic syndromes in patients undergoing
directional coronary atherectomy: in vivo evidence of thrombosis,
ulceration, and inflammation. Am Heart J 1994;128:419-426.[ISI][Medline]
- Uren NG,
Crake T, Lefroy DC, de Silva R, Davies GJ, Maseri A. Reduced coronary
vasodilator function in infarcted and normal myocardium after myocardial
infarction. N Engl J Med 1994;331:222-227.[Abstract/Full
Text]
- Gibson
CM, Ryan KA, Murphy SA, et al. Impaired coronary blood flow in nonculprit
arteries in the setting of acute myocardial infarction. J Am Coll Cardiol
1999;34:974-982.[ISI][Medline]
- Araujo
LI, Camici P, Spinks TJ, Jones T, Maseri A. Abnormalities in myocardial
metabolism in patients with unstable angina as assessed by positron
emission tomography. Cardiovasc Drugs Ther 1988;2:41-46.[Medline]
- Wallsh
E, Weinstein GS, Franzone A, Clavel A, Rossi PA, Kreps E. Inflammation of
the coronary arteries in patients with unstable angina. Tex Heart Inst J
1986;13:105-108.[ISI]
- Liuzzo
G, Biasucci LM, Gallimore JR, et al. The prognostic value of C-reactive
protein and serum amyloid A protein in severe unstable angina. N Engl J
Med 1994;331:417-424.[Abstract/Full
Text]
- Biasucci
LM, Vitelli A, Liuzzo G, et al. Elevated levels of interleukin-6 in
unstable angina. Circulation 1996;94:874-877.[Abstract/Full
Text]
- Liuzzo
G, Biasucci LM, Gallimore JR, et al. Enhanced inflammatory response in
patients with preinfarction unstable angina. J Am Coll Cardiol
1999;34:1696-1703.[ISI][Medline]
Edward E.
Rylander, M.D.
Diplomat American
Board of Family Practice.
Diplomat American
Board of Palliative Medicine.