Hypercoagulability Syndromes
Author Information
<http://archinte.ama-assn.org/issues/v161n20/rfull/#aainfo> Robert H.
Thomas, MD
Hypercoagulability can be defined as the tendency to have thrombosis as a
result of certain inherited and/or acquired molecular defects. Clinical
manifestations of hypercoagulability can be devastating and even lethal. In
the past 20 years, the origin of most of these diverse hypercoagulability
syndromes has been elucidated. Currently, hypercoagulability disorders can
be correctly diagnosed in approximately 80% to 90% of patients. Defining the
cause of hypercoagulability may determine the type and duration of treatment
for the associated thrombosis. The discovery of an occult carcinoma allows
for the possibility of early and possibly curative treatment. Finding a
genetic defect in coagulation allows for testing of asymptomatic family
members as well. The purpose of this review is to provide internists with a
logical approach to the identification and treatment of hypercoagulability
syndromes.
Arch Intern Med. 2001;161:2433-2439
IRA00051
Most commonly, thrombosis is the result of more than one "hit." For example,
patients with the factor V Leiden defect may be asymptomatic until they
start taking oral contraceptives. Patients with antithrombin deficiency may
go on without incident until they undergo a hernia repair. Also, multiple
genetic defects predispose one to thrombosis much more than does a single
defect. 1 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r1> , 2
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r2> Some defects are
known to be more powerful predictors than others. Therefore,
hypercoagulability is not a uniform disease process but rather a host of
predisposing conditions that may or may not be expressed as thrombosis,
depending on environmental insults and the strength and number of
predisposing factors.
INDIVIDUAL SYNDROMES
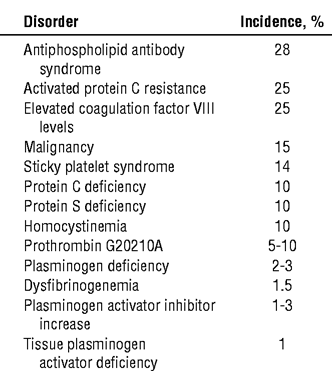
The following is a list of the disorders that cause hypercoagulability and
their approximate incidences. Since these were derived from different
studies, percentages cannot be exact. Also, incidences may vary, depending
on the ethnic backgrounds of persons in a particular geographic area.
Antiphospholipid Antibody Syndrome
The antiphospholipid antibody syndrome is probably the most common of the
hypercoagulable disorders. It is caused by a heterogeneous family of
immunoglobulins that bind to plasma proteins that have an affinity for
phospholipid surfaces. These antigens include B2 glycoprotein I,
prothrombin, high- and low-molecular-weight kininogens, annexin V, activated
protein C, and activated protein S. It is usually acquired and can be
divided into the lupus anticoagulant syndrome and the anticardiolipin
antibody syndrome. Both of these syndromes may be associated with other
disorders, such as collagen vascular diseases or infections, but are more
often primary. Antiphospholipid antibody syndrome can also be associated
with use of the following medications: phenytoin, quinidine, hydralazine,
procainamide hydrochloride, phenothiazines, interferon, cocaine, quinine,
and the combination product of pyrimethamine and sulfadoxine. Usually, a
patient will have one syndrome or the other but not both. Multiple
mechanisms as to the reason for hypercoagulability have been postulated, but
the exact cause is unknown at this time. The risk of thrombosis is 5.5% per
year for symptomatic patients. 3
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r3>
The lupus anticoagulant is directed against phospholipids, which then causes
an in vivo prolongation in the prothrombin time (PT), partial thromboplastin
time (PTT), or the Russell viper venom time. These values do not correct
with normal plasma. However, the addition of phospholipids will correct the
abnormality. Despite the prolonged coagulation times, thrombosis is the
predominant feature of this syndrome. The PT and PTT are not sensitive
enough to be used as a screening tool for the lupus anticoagulant. Instead,
the Russell viper venom time must be used. Venous thrombosis is much more
common than arterial thrombosis in these patients. 4
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r4>
The anticardiolipin antibody syndrome is 5 times more common than the lupus
anticoagulant syndrome. 5
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r5> Antibodies can be
detected by enzyme-linked immunosorbent assay. Both IgG and IgM are
associated with thrombosis. 4
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r4> , 5
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r5> A total of 1% to 7%
of asymptomatic individuals have low titers of these antibodies. 6
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r6> Even asymptomatic
persons have a 1% risk per year of thrombosis. This increases to 6% in those
with high titers. 7 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r7>
This contrasts with a 0.1% risk per year in the general population. Venous
and arterial thrombi are equally common. 4
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r4> Other
manifestations include valvular abnormalities, livedo reticularis,
superficial thrombophlebitis, ulcers, adrenal hemorrhage, fetal wastage,
chorea, transverse myelopathy, and thrombocytopenia.
The treatment of patients with antiphospholipid antibody syndrome who have
had thrombosis is long-term anticoagulation until the antibody has been
absent for at least 6 months. 8
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r8> The drug of choice
is low-molecular-weight heparin sodium since in 65% of patients warfarin
sodium therapy fails 8
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r8> and the
international normalized ratio is unreliable in monitoring the intensity of
therapy. If warfarin must be used, an international normalized ratio target
range of 3 to 4 should be sought. 9
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r9> The treatment of
fetal wastage syndrome is beyond the scope of this review. There is no clear
indication for therapy in asymptomatic persons; however, aspirin therapy
would be reasonable in this population because the risk of thrombosis is
higher than normal. Other treatments, such as corticosteroids,
cyclophosphamide, and plasma exchange, have been used for severely
symptomatic disease, but their roles in routine management are not well
established.
Activated Protein C Resistance
Activated protein C resistance (eg, factor V Leiden) is the most common
inherited disorder that causes hypercoagulability. Factor V Leiden is
present in 5% of whites but virtually absent in Africans and Asians.
However, 1% of African Americans have the mutation, reflecting racial
mixing. 10 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r10> It
results from a point mutation in the factor V gene, which causes the
substitution of glutamine for arginine at position 506. 11
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r11> (Several other
rare factor V gene mutations that can lead to activated protein C resistance
have also been described. 12-14
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r12> ) Consequently, 1
of 3 activated protein C cleavage sites is lost. The result is an impaired
inactivation of factor V by activated protein C. Venous thromboses and fetal
wastage may occur. It is not an important risk factor for arterial disease
except in the presence of smoking or other known risk factors. 15
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r15> , 16
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r16> Those with factor
V Leiden have a 2- to 3-fold risk for venous thrombosis compared with
healthy subjects. The risk in homozygotes is 80-fold. 17
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r17> Heterozygous
factor V Leiden is, therefore, a relatively mild risk factor for thrombosis.
15 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r15> , 18
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r18> The annual rate of
thrombosis is 0.28%. 19
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r19> Six percent of
patients will have a thrombosis by the age of 65 years. 20
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r20> Sixty percent of
patients who experience thrombosis have a predisposing event, such as oral
contraceptive use or pregnancy. 21
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r21> The presence of
this mutation does not appear to affect life expectancy, and many patients
will remain asymptomatic. Therefore, patients with no history of thrombosis
should not be treated prophylactically with long-term anticoagulation. 22
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r22> Functional tests
for activated protein C resistance should be used to screen for the
disorder, and positive results should be confirmed with polymerase chain
reaction for the genetic mutation. However, patients with phenotypic
resistance to activated protein C have an increased risk of thrombosis even
if it is not due to factor V Leiden. 20
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r20> Functional tests
may still be performed while patients undergo anticoagulation.
Elevated Coagulation Factor VIII Levels
Elevated coagulation factor VIII levels appear to be nearly as common a risk
factor for thrombosis as factor V Leiden. The Leiden Thrombophilia Study
found an 11% incidence in healthy controls and a 25% incidence in patients
with venous thrombosis. The odds ratio for thrombosis was 4.8 for subjects
with levels greater than 150 IU/dL vs those with levels less than 100 IU/dL.
23 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r23> , 24
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r24> For every 10-IU/dL
rise in levels, the risk for a single episode of deep venous thrombosis
(DVT) increases 10% and the risk for recurrent DVT increases 24%. 25
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r25> Levels of
coagulation factor VIII are not elevated because of the acute-phase reaction
but appear to be constitutively increased in most patients with thrombosis,
since coagulation factor VIII levels are elevated independently of
C-reactive protein and fibrinogen, and 94% of patients continue to have high
levels throughout long-term follow-up. 26
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r26> , 27
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r27> Pregnancy and oral
contraceptive use may also raise levels. The use of oral contraceptives in
patients with increased coagulation factor VIII levels raises the risk of
thrombosis 10-fold over patients with neither risk factor. 28
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r28> The genetic basis
for increased coagulation factor VIII levels is not well understood at this
time; however, one small study 25
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r25> showed high
concordance rates for first-degree adult family members.
Malignancy
Cancer is the second most common acquired cause of hypercoagulability,
accounting for 10% to 20% of spontaneous DVTs. Indeed, 15% of patients with
cancer have clinical thromboses and about 50% have thromboses on autopsy. 29
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r29> Cancer not only
causes hypercoagulability but may also produce endothelial injury and venous
stasis. Hypercoagulability is especially frequent in mucin-secreting
adenocarcinomas, brain tumors, acute promyelocytic leukemia, and
myeloproliferative disorders.
Arterial thrombosis is much less common than venous thrombosis and is most
often the result of nonbacterial thrombotic endocarditis or disseminated
intravascular coagulation. Ninety percent of patients with cancer have
clotting abnormalities, such as increased fibrinogen, clotting factors,
fibrin degradation products, and platelets. 30
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r30> , 31
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r31> Overt disseminated
intravascular coagulation is rare. There is no consensus as to the value of
measuring coagulation markers in predicting thrombosis in individual
patients with cancer.
Some cancers underlying spontaneous DVT are occult, early stage, and
curable. However, there is no proof that aggressive diagnostic testing leads
to improvement in survival. Most experts recommend a thorough history and
physical examination, routine blood tests, chest x-ray examination,
urinalysis, and age- and sex-specific screening, such as prostate-specific
antigen, Papanicolaou smear, lower endoscopy, mammography, and fecal occult
blood testing. Suspicious findings should be aggressively evaluated. In
addition, patients without evidence of cancer should be followed up closely
for the ensuing 2 years, during which time virtually all occult cancers will
become clinically apparent.
The initial treatment of thromboses is the same as in patients without
cancer. However, treatment should be continued indefinitely until the
patient is cured of the malignancy and is no longer receiving chemotherapy.
If anticoagulation is contraindicated as with cerebral or pericardial
metastases, primary brain tumors, or severe thrombocytopenia, an inferior
vena cava filter may be placed. Long-term treatment may be with
low-molecular-weight heparin or warfarin, although anecdotal evidence
suggests that heparin may lead to fewer thrombotic recurrences than
warfarin. 32 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r32>
Certainly, warfarin failure should lead to a switch to heparin. If heparin
fails, an inferior vena cava filter should then be placed. Thrombolytic
agents should only be used in patients with cancer who have a good prognosis
and either pulmonary embolism with hemodynamic compromise or severe
iliofemoral thrombosis of less than 4 days' duration. Of course, aggressive
DVT prophylaxis with low-dose subcutaneous heparin or low-molecular-weight
heparin (depending on severity and number of risk factors) should be carried
out in patients with cancer who are hospitalized, immobilized, or undergoing
surgery.
Sticky Platelet Syndrome
The sticky platelet syndrome is an autosomal dominant disorder that results
in platelets that are hyperaggregable to epinephrine and/or adenosine
diphosphate. Venous or arterial thrombosis may occur. 33
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r33> Episodes are more
common during emotional stress. Retinal vascular thrombosis appears to be
associated with this entity. Fetal wastage may also occur. It is diagnosed
with platelet aggregation studies. Treatment is with low-dose aspirin (81
mg). If platelet aggregability does not normalize, aspirin, 325 mg, may be
tried. 34 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r34> If there
is still no response, then clopidogrel (an adenosine diphosphate receptor
antagonist similar to but better tolerated than ticlopidine hydrochloride)
may be used.
Protein C Deficiency
Protein C deficiency is an autosomal dominant trait that may be caused by a
decrease in absolute levels of protein C or a decrease in its function.
Deficiency of protein C occurs in 1 of 250 controls. 35
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r35> Protein C is made
in the liver and is vitamin K dependent. It acts to inactivate factor V and
factor VIII:C. It requires factor S as a cofactor and is activated by
thrombin, when thrombin is bound to thrombomodulin.
In families with thromboses and protein C deficiency, thromboses begin in
the late teens. 36 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r36>
Seventy-five percent of affected individuals will have 1 or more events. 38
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r38> The relative risk
is 7.3. 18 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r18> The
annual incidence is 1%. 19
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r19> Seventy percent of
episodes are spontaneous. 37
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r37> Both DVT and
pulmonary embolism are the most common manifestations. Superficial
thrombophlebitis is also common. 38
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r38> Arterial events
are rare.
The optimal time to investigate is at least 10 days after warfarin therapy
is stopped, since both warfarin and acute thrombosis decrease protein C
levels. Levels below 55% of normal are likely to be genetically deficient;
55% to 65% is borderline. Abnormal results should always be repeated for
confirmation and family studies performed. 39
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r39>
The short-term management of thrombosis is with heparin or
low-molecular-weight heparin. Warfarin may be used for long-term treatment;
however, doses should be started low and titrated upward slowly only after
heparin is therapeutic because of the risk of warfarin necrosis. 40
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r40> In fact, one third
of patients with warfarin necrosis have an underlying protein C deficiency.
Protein S Deficiency
Protein S is vitamin K dependent and is synthesized by hepatocytes and
megakaryocytes. It acts as a cofactor for protein C. Fifty percent
circulates free and 50% circulates bound to C4b binding protein. Deficiency
is transmitted autosomally dominant and can be quantitative or qualitative.
Seventy-four percent of patients develop DVT; 72% develop superficial
thrombophlebitis. 41
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r41> The relative risk
of thrombosis is 8.5. 18
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r18> The annual
incidence is 1% 19 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r19>
; 56% of episodes are spontaneous. Arterial events are uncommon. One half of
patients who develop thromboses do so by the age of 25 years. 41
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r41>
Short-term therapy is standard. Long-term therapy is with warfarin or
low-molecular-weight heparin. Since warfarin necrosis may occur, therapy
should be started with warfarin at low doses and increased slowly after
heparin has been administered. While the patient is undergoing warfarin
therapy, protein C and S levels decrease by 50% within 48 hours and then
increase to 70% of usual levels after 2 weeks. Therefore, levels below 60%
of normal while taking warfarin in the long term are suspicious for
deficiency. 8 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r8>
Homocystinemia
Elevated levels of homocysteine are known to be a risk factor for arterial
and venous thrombosis and fetal wastage. Homocysteine is an intermediate of
methionine metabolism and, therefore, elevated levels may result from
cystathionine beta-synthase deficiency, homozygous expression of the
thermolabile form of methylenetetrahydrofolate reductase, or from B12 or
folic acid deficiency. Mild-to-moderate increases in homocysteine occur in
5% to 10% of the population. 42
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r42> The relative risk
of thrombosis is 2.6. 43
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r43>
Elevated homocysteine levels are thought to cause thromboses via several
mechanisms, including (1) decreased protein C activation, (2) increased
factor V activity, (3) induction of endothelial cell tissue factor activity,
(4) inhibition of thrombomodulin expression and activation, (5) decreased
antithrombin activity, and (6) enhanced affinity of lipoprotein(a) and
fibrin. 44-46 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r44>
Measurement of homocysteine levels is not well standardized, and acute
thrombosis may raise homocysteine levels. Dietary supplementation with
vitamin B6, B12, and folic acid can lower homocysteine levels. 47
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r47> However, reduction
of homocysteine levels has not been shown to reduce thrombotic
complications. Folate supplementation (400 µg/d) may decrease levels by 30%
to 42%. B12 supplementation (100 µg/d) may decrease levels by 15%. B6
supplementation (3 µg/d) only reduces levels if there is a preexisting
deficiency. Thrombosis is treated in standard fashion in addition to vitamin
supplementation.
Antithrombin Deficiency
Antithrombin is made in the liver and endothelial cells. It inactivates
thrombin and other serine proteases. Deficiency is an autosomal dominant
disorder and occurs in 1 of 5000 healthy blood donors. 48
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r48> The protein may be
absent or dysfunctional. The normal concentration is 150 µg/mL. Thrombosis
may occur at less than 75% of this amount. Patients may present with DVT or
pulmonary embolism. Mesenteric vessels appear to be particularly
susceptible. Arterial events are rare. Fifty percent of patients are
asymptomatic. Thromboses occur early in life, with two thirds of patients
presenting by the age of 35 years. Forty percent of thromboses are
spontaneous. 49 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r49>
The relative risk of thrombosis is 8.1, 18
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r18> and the annual
incidence of thrombosis is 1%. 19
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r19>
Acute thrombosis, heparin, and other systemic diseases may decrease
antithrombin levels. 8
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r8> Warfarin may raise
deficient levels into the normal range. 50
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r50> Therefore, low
levels in a patient during acute thrombosis or while taking heparin should
be confirmed when the patient is not undergoing therapy. Likewise, normal
levels while the patient is taking warfarin should be confirmed when the
patient is not undergoing therapy.
Treatment of acute thrombosis is with low-molecular-weight heparin because
deficiency may cause resistance to unfractionated heparin. 51
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r51> In fact, heparin
resistance may be a clue to the presence of this deficiency. Lifelong
therapy should be considered for spontaneous or recurrent thromboses.
Prophylactic treatment of asymptomatic individuals is controversial but
usually is limited to high-risk situations, such as pregnancy or surgery.
Antithrombin concentrate may be considered for situations in which both
thrombosis and bleeding may occur, such as labor and delivery, where
anticoagulation might be contraindicated. 52
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r52>
Dysfibrinolysis
There are 5 major forms of dysfibrinolysis: (1) congenital plasminogen
deficiency, (2) tissue plasminogen activator deficiency, (3) increased
plasminogen activator inhibitor, (4) congenital dysfibrinogenemia, and (5)
factor XII deficiency. Long-term treatment may be with warfarin or
low-molecular-weight heparin for all patients.
Congenital plasminogen deficiency is a rare autosomal dominant disorder
caused by either absent or dysfunctional plasminogen. Clinically, it mimics
protein C and S deficiencies. Symptoms usually begin in the late teens. Most
commonly, it presents with DVT or pulmonary embolism. Arterial events are
uncommon. Events usually occur when plasminogen levels are less than 40% of
the normal values. The results of routine coagulation studies are normal. 53
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r53> Treatment is
standard.
Congenital deficiency of tissue plasminogen activator and congenital
increases of plasminogen activator inhibitor are exceedingly rare. Acquired
abnormalities are more common. They may occur with diabetes mellitus,
inflammatory bowel disease, and coronary atherosclerosis. 54-56
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r54>
Most congenital dysfibrinogenemias occur in asymptomatic individuals (55% of
patients) or cause mild hemorrhagic disorders (20%). Only 20% are associated
with thrombosis. 57 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r57>
Venous thrombosis is most common but arterial events may occur. They are
usually autosomal dominant. They may be detected with abnormal thrombin
times or reptilase clotting times. Treatment of thrombosis consists of
heparin or low-molecular-weight heparin followed by warfarin.
Factor XII deficiency is inherited in autosomal dominant fashion. It is
involved in plasmin generation. Thus, patients will have a prolonged PTT,
yet have a thrombotic diathesis. Arterial and venous thromboses and fetal
wastage are common. Approximately 8% of deficient subjects develop
thromboses. 58 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r58>
Factor XII deficiency should be suspected when a patient with thrombosis has
a prolonged PTT that corrects with the addition of normal plasma. A factor
XII assay should then be performed. Treatment is with low-molecular-weight
heparin followed by warfarin or continuation of low-molecular-weight
heparin. Standard unfractionated heparin should not be used because of
difficulties in monitoring the PTT.
Prothrombin G20210A
Prothrombin G20210A mutation is a relatively recently discovered defect in
which there is a G to A transition at nucleotide position 20210. This
mutation increases prothrombin activity and levels. 59
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r59> It is found in
2.3% of healthy controls. The incidence is twice as high in people from
southern Europe than from northern Europe, and it is rare in Africans and
Asians. 60 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r60> It may
be detected through DNA analysis. At this time, it must be considered a very
mild risk factor for venous and arterial thrombosis. The relative risk is
approximately 2 to 3 times that of individuals without the mutation. 61-63
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r61>
Other Hypercoagulable Syndromes
Heparin cofactor II inhibits thrombin by mimicking the cleavage sites of
thrombin and forming a stable complex with it, thus acting as a "suicide"
substrate. Deficiency is rare and could theoretically cause thrombotic
potential, but its exact role is controversial. Heparin is effective in the
presence of heparin cofactor II deficiency.
Tissue factor pathway inhibitor is a plasma component that binds and
inhibits factor Xa directly. This complex then binds to the tissue
factorfactor VIIa complex, blocking its activity as well. Unstimulated
plasma levels do not appear to be related to thrombosis. However, plasma
levels measured 10 minutes after intravenous heparin, 7500 U, is
administered correlate with venous thrombosis. 64
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r64> The role of tissue
factor pathway inhibitor and its incidence in thrombophilia are currently
unknown.
Thrombomodulin mutations have also been implicated in thrombophilia but
prevalence and degree of risk are unknown. 65
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r65>
The Wein-Penzing defect is an extremely rare deficiency of the lipoxygenase
metabolic pathway that results in the compensatory increase of the
cyclooxygenase pathway and, therefore, elevated thromboxane levels. Thus,
platelets are in a state of increased activation.
INVESTIGATION OF HYPERCOAGULABILITY
Various clinical features should suggest hypercoagulability, including
thrombosis at an early age (<50 years), family history of thrombosis,
recurrent idiopathic thrombosis, thrombosis at an unusual site (except for
effort-related upper extremity DVT 66
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r66> ), spontaneous
thrombosis or only mild provocation, unexplained spontaneous abortions,
massive thrombosis, and warfarin-induced skin necrosis. Information from the
history and physical examination determines the likelihood of the underlying
disorder. For example, a young patient who presents with a strong family
history of thrombosis suggests a genetic disorder. A patient with systemic
lupus erythematosus is likely to have the antiphospholipid antibody
syndrome. An older patient with weight loss, early satiety, and epigastric
pain is likely to have a gastric carcinoma.
All patients should have a complete blood cell count performed, including
platelets, to exclude myeloproliferative disorders. Abnormalities in PT and
PTT suggest either the lupus anticoagulant or factor XII deficiency.
Antiphospholipid antibodies should be obtained. Screening for cancer as
outlined earlier should be performed. Patients in whom this evaluation is
negative and all patients with a positive family history of thrombosis
should undergo testing for the common genetic disorders ( Table 1
<http://archinte.ama-assn.org/issues/v161n20/fig_tab/ira00051_t1.html> ).
Table 2
<http://archinte.ama-assn.org/issues/v161n20/fig_tab/ira00051_t2.html>
provides a list of the approximate costs of the various tests for
hypercoagulability. Laboratory investigation for these disorders is
generally unreliable during acute thrombosis and while undergoing
anticoagulant therapy. Thus, studies are optimally performed while the
patient is not taking anticoagulants and is in the asymptomatic state. If
tests are performed while the patient is taking anticoagulants, knowledge of
the alteration of the individual factors by the specific anticoagulant is
essential. Depending on the level of suspicion for a genetic defect,
referral to a hematologist for testing of the rarer defects may be indicated
if the prior workup is unrewarding. A useful mnemonic for the common causes
of hypercoagulability is CALMSHAPES: protein C deficiency, Antiphospholipid
antibody syndrome, factor V Leiden; Malignancy, protein S deficiency,
Homocystinemia, Antithrombin deficiency, Prothrombin G20210A, increased
factor VIII (Eight), Sticky platelet syndrome.
TREATMENT
When considering a patient for indefinite therapy, many factors must be
considered: (1) the number, site, and severity of thromboses; (2)
spontaneous vs provoked thrombus; (3) the sex and lifestyle of the patient;
(4) the strength of the predictive value for thrombosis of the particular
hypercoagulable disorder; (5) the compliance of the patient; and (6) the
patient's personal value construct.
Few guidelines exist for indefinite therapy in hypercoagulable patients. A
recent study 67 <http://archinte.ama-assn.org/issues/v161n20/rfull/#r67>
that favored indefinite therapy in anyone with an unprovoked thrombosis was
terminated prematurely. Bauer 68
<http://archinte.ama-assn.org/issues/v161n20/rfull/#r68> divides patients
with hereditary defects into 2 groups: high risk (2 spontaneous episodes, 1
spontaneous life-threatening thrombosis, 1 thrombosis at an unusual site, or
1 thrombosis in the presence of >1 defect) and moderate risk (asymptomatic
individuals or 1 thrombosis in response to a prothrombotic stimulus). In the
high-risk group, he recommends indefinite anticoagulation. In the
moderate-risk group, he recommends vigorous prophylaxis only for high-risk
situations. Since no long-term studies have been performed comparing
lifetime anticoagulation treatment with short-term anticoagulation therapy,
definitive recommendations cannot be made at this time. Until these studies
are performed, Figure 1
<http://archinte.ama-assn.org/issues/v161n20/fig_tab/ira00051_f1.html> may
be used as a guide to the evaluation and management of hypercoagulable
disorders.
CONCLUSIONS
The evaluation and treatment of a patient suspected of having
hypercoagulability cannot be generalized at this time. The clinician must
consider many patient factors with statistical probabilities to determine
what conditions should be investigated. When a hypercoagulable syndrome is
diagnosed, further judgment must be exercised to then decide the best course
of treatment. Oversimplifications on the evaluation and treatment of
hypercoagulable syndromes are not helpful and may result in harm to
individual patients.
Author/Article Information
From the Department of General Medicine, University of Miami School of
Medicine, Miami, Fla.
Corresponding author and reprints: Robert H. Thomas, MD, Department of
General Medicine, University of Miami School of Medicine, 1475 NW 12th Ave,
Third Floor, Miami, FL 33136 (e-mail: [log in to unmask]
<mailto:[log in to unmask]> ).
Accepted for publication April 9, 2001.
I would like to thank Barry Materson, MD, for his excellent advice and
assistance in editing the manuscript.
REFERENCES
1. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr1>
Salomon O, Steinberg DM, Zivelin A, et al.
Single and combined prothrombotic factors in patients with idiopathic venous
thromboembolism: prevalence and risk assessment.
Arterioscler Thromb Vasc Biol.
1999;19:511-518.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10073951>
2. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr2>
DeStefano U, Martinelli I, Mannucci P, et al.
The risk of recurrent deep venous thrombosis among heterozygous carriers of
both factor V Leiden and the G20210A prothrombin mutation.
N Engl J Med.
1999;341:801-806.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10477778>
3. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr3>
Finazzi G, Brancaccio U, Moia M, et al.
Natural history and risk factors for thrombosis in 360 patients with
antiphospholipid antibodies: a four-year prospective study from the Italian
Registry.
Am J Med.
1996;100:530-536.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8644765>
4. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr4>
Bick RL.
Antiphospholipid thrombosis syndromes: etiology, pathophysiology, diagnosis
and management.
Int J Hematol.
1997;65:193-213.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9114592>
5. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr5>
Bick RL.
The antiphospholipid thrombosis syndromes: fact, fiction, confusion and
controversy.
Am J Clin Pathol.
1993;100:477-480.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8249884>
6. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr6>
Petri M.
Epidemiology of the antiphospholipid syndrome.
In: Asherson RA, Cervera R, Piette JC, Schoenfeld Y, eds. The
Antiphospholipid Syndrome. Boca Raton, Fla: CRC Press; 1996:13-28.
7. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr7>
Bick RL, Jakway J, Baker WF Jr.
Deep vein thrombosis: prevalence of etiologic factors and results of
management in 100 consecutive patients.
Semin Thromb Hemost.
1992;18:267-274.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
1631576>
8. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr8>
Bick RL, Kaplan H.
Syndromes of thrombosis and hypercoagulability.
Med Clin North Am.
1998;82:409-458.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9646773>
9. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr9>
Moll S, Ortel TL.
Monitoring warfarin therapy in patients with lupus anticoagulants.
Ann Intern Med.
1997;127:177-185.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9245222>
10. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr10>
DeStefano U, Chirrolo P, Paciaroni K, et al.
Epidemiology of factor V Leiden: clinical implications.
Semin Thromb Hemost.
1998;24:367-379.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9763354>
11. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr11>
Dahlback B.
New molecular insights into the genetics of thrombophilia: resistance to
activated protein C caused by Arg 506 to Gln mutation in factor V as a
pathogenic risk factor for venous thrombosis.
Thromb Haemost.
1995;74:139-148.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8578447>
12. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr12>
Chan WP, Lee CK, Kwong YL, et al.
A novel mutation of Arg 306 of factor V gene in Hong Kong Chinese.
Blood.
1998;91:1135-1139.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9454741>
13. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr13>
Williamson D, Brown K, Luddington R, et al.
Factor V Cambridge: a new mutation (Arg 306 to Thr) associated with
resistance to activated protein C.
Blood.
1998;91:1140-1144.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9454742>
14. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr14>
Alhenc-Gelas M, Nicaud V, Gandrille S, et al.
The factor V gene A4070G mutation and the risk of venous thrombosis.
Thromb Haemost.
1999;81:193-197.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10063990>
15. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr15>
Ridker PM, Hennekens CH, Lindpainter K, et al.
Mutation in the gene coding for coagulation factor V and the risk of
myocardial infarction, stroke and venous thrombosis in apparently healthy
young men.
N Engl J Med.
1995;332:912-917.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
7877648>
16. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr16>
Siscovick DS, Schwartz SM, Rosendaal FR, et al.
Thrombosis in the young: effect of atherosclerotic risk factors on the risk
of myocardial infarction associated with prothrombotic factors.
Thromb Haemost.
1997;78:7-12.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9198120>
17. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr17>
Rosendaal FR, Koster T, Vandenbroucke JP, et al.
High risk of thrombosis in patients homozygous for factor V Leiden.
Blood.
1995 Mar 15;85(6):1504-1508.
18. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr18>
Martinelli I, Mannucci PM, DeStefano V, et al.
Different risks of thrombosis in four coagulation defects associated with
inherited thrombophilia: a study of 150 families.
Blood.
1998;92:2353-2358.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9746774>
19. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr19>
Simioni P, Sanson BJ, Prandoni P, et al.
Incidence of venous thromboembolism in families with inherited
thrombophilia.
Thromb Haemost.
1999;81:198-202.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10063991>
20. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr20>
Rodeghiero F, Tosetto A.
Activated protein C resistance and factor V Leiden mutation are independent
risk factors for venous thromboembolism.
Ann Intern Med.
1999;130:643-650.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10215560>
21. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr21>
Faioni EM, Razzari C, Martinelli I, et al.
Resistance to activated protein C in unselected patients with arterial and
venous thrombosis.
Am J Hematol.
1997;55:59-64.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9208999>
22. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr22>
Hille ET, Westenderp RG, Vandenbroucke JP, et al.
Mortality and causes of death in families with the factor V Leiden mutation
(resistance to activated protein C).
Blood.
1997;89:1963-1967.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9058717>
23. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr23>
van der Meer FJM, Koster T, Vandenbroucke JP, et al.
The Leiden Thrombophilia Study.
Thromb Haemost.
1997;78:631-635.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9198229>
24. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr24>
Koster T, Blann AD, Briet E, et al.
Role of clotting factor VIII in effect of von Willebrand factor on
occurrence of deep vein thrombosis.
Lancet.
1995;345:152-155.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
7823669>
25. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr25>
Kraaijenhagen R, Pieternella S, Koopman M, et al.
High plasma concentrations of factor VIII:C is a major risk factor for
venous thromboembolism.
Thromb Haemost.
2000;83:5-9.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10669145>
26. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr26>
O'Donnell J, Tuddenham EGD, Manning R, et al.
High prevalence of elevated factor VIII levels in patients referred for
thrombophilia screening: role of increased synthesis and relationship to the
acute phase reaction.
Thromb Haemost.
1997;77:825-828.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9184386>
27. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr27>
O'Donnell J, Mumford A, Manning R, et al.
Elevation of FVIII:C in venous thromboembolism is persistent and independent
of the acute phase response.
Thromb Haemost.
2000;83:10-13.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10669146>
28. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr28>
Bloemenkamp KWM, Helmerhorst FM, Rosendaal FR, Vandenbroucke JP.
Venous thrombosis, oral contraceptives and high factor VIII levels.
Thromb Haemost.
1999;82:1024-1027.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10494758>
29. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr29>
Luzzato G, Schafer AI.
The prethrombotic state in cancer.
Semin Oncol.
1990;17:147-159.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2183357>
30. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr30>
Rickles FR, Levine M, Edwards RL.
Hemostatic alterations in cancer patients.
Cancer Metastasis Rev.
1992;11:237-248.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
1423816>
31. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr31>
Sun NC, McAfee WM, Hum GJ, et al.
Hemostatic abnormalities in malignancy, a prospective study in one hundred
eight patients, part I: coagulation study.
Am J Clin Pathol.
1979;71:10-16.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
420161>
32. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr32>
Sack GH, Levin J, Bell W.
Trousseau's syndrome and other manifestations of chronic disseminated
coagulopathy in patients with neoplasms: clinical, pathologic, and
therapeutic features.
Medicine (Baltimore).
1977;56:1-37.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
834136>
33. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr33>
Mammen EF.
Sticky platelet syndrome.
Semin Thromb Hemost.
1999;25:361-365.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10548069>
34. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr34>
Mammen EF.
Ten years' experience with the "sticky platelet syndrome.".
Clin Appl Thromb Hemost.
1995;1:66-72.
35. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr35>
Tait RC, Walker ID, Reitsma PH, et al.
Prevalence of protein C deficiency in the healthy population.
Thromb Haemost.
1995;73:87-93.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
7740502>
36. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr36>
Griffin JH.
Clinical studies on protein C.
Semin Thromb Hemost.
1984;10:162-166.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
6377499>
37. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr37>
Broekmans AW, Veltkamp JJ, Bertina RM.
Congenital protein C deficiency and venous thromboembolism: a study of three
Dutch families.
N Engl J Med.
1983;309:340-344.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
6688122>
38. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr38>
Broekmans AW.
Hereditary protein C deficiency.
Haemostasis.
1985;15(4):233-240.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
3840112>
39. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr39>
Bauer K.
Hypercoagulable states.
In: Hoffman R, Furie B, Cohen H, Benz E Jr, Silberstein L, Shattil S, eds.
Hematology: Basic Principles and Practice. 2nd ed. Camden Town, London,
England: Churchill Livingstone Inc; 1995:1781-1794.
40. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr40>
Baker W Jr, Bick R.
Treatment of hereditary and acquired thrombophilic disorders.
Semin Thromb Hemost.
1999;25:387-406.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10548072>
41. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr41>
Engesser L, Broekmans AW, Briet E, et al.
Hereditary protein S deficiency: clinical manifestations.
Ann Intern Med.
1987;106:677-682.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2952034>
42. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr42>
VanCott EM, LaPosata M.
Laboratory evaluation of hypercoagulable states.
Hematol Oncol Clin North Am.
1998;12:1141-1166.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9922930>
43. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr43>
den Heijer M, Rosendaal FR, Blom HJ, Gerrits WB, Bos GM.
Hyperhomocysteinemia and venous thrombosis: a meta-analysis.
Thromb Haemost.
1998;80:874-877.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9869152>
44. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr44>
Rees MM, Rodgers GM.
Homocysteinemia: association of a metabolic disorder with vascular disease
and thrombosis.
Thromb Res.
1993;71:337-359.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8236162>
45. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr45>
Harpel PC.
Homocysteine, atherogenesis and thrombosis.
Fibrinolysis.
1997;11:77-80.
46. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr46>
Bellamy MF, McDowell IFW.
Putative mechanisms for vascular damage by homocysteine.
J Inherit Metab Dis.
1997;20:307-315.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9211203>
47. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr47>
Franken DG, Boers GHJ, Blom HJ, et al.
Treatment of mild hyperhomocysteinemia in vascular disease patients.
Arterioscler Thromb.
1994;14:465-470.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8123653>
48. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr48>
Tait RC, Walker ID, Perry DJ, et al.
Prevalence of antithrombin deficiency in the healthy population.
Br J Haematol.
1994;87:106-112.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
7947234>
49. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr49>
Thaler E, Lechner K.
Antithrombin III deficiency and thromboembolism.
Clin Haematol.
1981;10:369-390.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
7032781>
50. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr50>
Marciniak E, Farley CH, DeSimone PA.
Familial thrombosis due to antithrombin III deficiency.
Blood.
1974;43:219-231.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
4810073>
51. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr51>
Francis J.
Laboratory investigation of hypercoagulability.
Semin Thromb Hemost.
1998;24:111-126.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9579632>
52. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr52>
Menache D.
Replacement therapy in patients with hereditary antithrombin III deficiency.
Semin Hematol.
1991;28:31-38.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2017690>
53. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr53>
Mammen EF.
Plasminogen abnormalities.
Semin Thromb Hemost.
1983;9:50-51.
54. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr54>
Kirschsteln W, Simianer S, Dempfle CE.
Impaired fibrinolytic capacity and tissue plasminogen activator release in
patients with restenosis after PTCA.
Thromb Haemost.
1989;62:772-775.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2510351>
55. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr55>
DeJong E, Porte RJ, Knot EA.
Disturbed fibrinolysis in patients with inflammatory bowel disease.
Gut.
1989;30:188-194.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2495239>
56. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr56>
Juhan-Ungue I, Roul C, Alessi MC, et al.
Increased plasminogen activator inhibitor activity in non-insulin dependent
diabetic patientsrelationship with plasma insulin.
Thromb Haemost.
1989;61:370-373.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
2678583>
57. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr57>
Ebert R.
Index of Variant Human Fibrinogens.
Boca Raton, Fla: CRC Press; 1994.
58. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr58>
Goodnough LT, Saito H, Ratnoff OD.
Thrombosis or myocardial infarction in congenital clotting factor
abnormalities and thrombocytopenias: a report of 21 patients and a review of
previously reported cases.
Medicine (Baltimore).
1983;62:248-255.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
6348471>
59. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr59>
Simioni P, Tormene D, Manfrin D, et al.
Prothrombin antigen levels in symptomatic and asymptomatic carriers of the
20210A prothrombin variant.
Br J Haematol.
1998;103:1045-1050.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9886317>
60. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr60>
Rosendaal FR, Doggen CJ, Zivelin A, et al.
Geographic distribution of the 20210 G to A prothrombin variant.
Thromb Haemost.
1998;79:706-708.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9569177>
61. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr61>
Ridker PM, Hennekens CH, Miletich JP.
G20210A mutation in prothrombin gene and risk of MI, stroke, and venous
thrombosis in a large cohort of US men.
Circulation.
1999;99:999-1004.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10051291>
62. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr62>
Brown K, Luddington R, Williamson D, et al.
Risk of venous thromboembolism associated with a G to A transition at
position 20210 in the 3'-untranslated region of the prothrombin gene.
Br J Haematol.
1997;98:907-909.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9326187>
63. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr63>
Hillarp A, Zoller B, Svensson PJ, et al.
The 20210A allele of the prothrombin gene is a common risk factor among
Swedish outpatients with verified deep venous thrombosis.
Thromb Haemost.
1997;78:990-992.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9308741>
64. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr64>
Ariens R, Alberio G, Moia M, Mannucci P.
Low levels of heparin-releasable tissue factor pathway inhibitor in young
patients with thrombosis.
Thromb Haemost.
1999;81:203-207.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10063992>
65. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr65>
Doggen CJ, Kunz G, Rosendaal FR, et al.
A mutation in the thrombomodulin gene, 127G to A coding for Ala25Thr, and
the risk of myocardial infarction in men.
Thromb Haemost.
1998;80:743-748.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
9843165>
66. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr66>
Heron E, Lozinguez O, Alhenc-Gelas M, et al.
Hypercoagulable states in primary upper extremity DVT.
Arch Intern Med.
2000;160:382-386.
ABSTRACT <http://archinte.ama-assn.org/issues/v160n3/abs/ioi80780.html> |
FULL TEXT <http://archinte.ama-assn.org/issues/v160n3/rfull/ioi80780.html>
| PDF <http://archinte.ama-assn.org/issues/v160n3/rpdf/ioi80780.pdf> |
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10668841>
67. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr67>
Kearon C, Gent M, Hirsh J, et al.
A comparison of three months of anticoagulation with extended
anticoagulation for a first episode of idiopathic venous thromboembolism.
N Engl J Med.
1999;340:901-907.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
10089183>
68. <http://archinte.ama-assn.org/issues/v161n20/rfull/#rr68>
Bauer KA.
Management of patients with hereditary defects predisposing to thrombosis
including pregnant women.
Thromb Haemost.
1995;74:94-100.
MEDLINE
<http://www.ncbi.nlm.nih.gov/htbin-post/Entrez/query?db=m&form=6&Dopt=r&uid=
8578533>
Edward E. Rylander, M.D.
Diplomat American Board of Family Practice.
Diplomat American Board of Palliative Medicine.
|













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}